







Poikilodermatous mycosis fungoides (MF) is a rare clinical variant of patch-stage MF and is characterized by deep-red or brownish plaques with scaling, mottled dyspigmentation, atrophy, and telangiectasia affecting the trunk and extremities [1]. We have recently seen a patient with 34 years’ duration of the poikilodermatous lesion. To the best of my knowledge, this is the longest duration of the poikilodermatous lesion reported in the English literature.
A 68-year-old male was referred to us for evaluation of a 34-year history of widespread poikilodermatous patches over the trunk and extremities. He was a Chinese-Vietnam War veteran, and noticed the first appearance of scaly erythematous patches on his chest 34 years before. Later, it had gradually turned into poikiloderma and had progressed to involve the whole trunk and extremities over a period of 2 years. Similarly, his chin and lateral cheeks had been affected since 1 year earlier. This caused much cosmetic embarrassment to him. Before this, he had not received any medical treatment, although he had experienced slight itching. On examination, generalized, erythematous and poikilodermatous patches and thin plaques covered 80% of the body surface, including the trunk, flexural surfaces and extremities. The lesions were typically atrophic, telangiectatic, and intermingled with mottled hyperpigmentation and hypopigmentation (Figure 1 A). Whole-body computed tomography (CT) scanning revealed no evidence of lymph node or visceral involvement. A lesional skin biopsy showed necrotic keratinocytes and focal epidermotropism of atypical lymphocytes without spongiosis. There were vacuolar alterations of the basal layer, scattered melanophages, intermittent fibrosis of the collagen and lymphocytic infiltration in the papillary dermis (Figures 1 B and C). Immunohistochemically, the epidermotropic lymphocytes were positive for CD3, CD4 and CD45RO but were negative for CD8, CD20, CD79, TIA-1 and granzyme B (Figure 2). Gene rearrangement analysis showed the presence of monoclonal-type T cells. The results of blood tests for cell counts and the chemistry panel were within normal limits. Based on the aforementioned findings, poikilodermatous MF was diagnosed. The patient was treated with narrow-band UVB, and his skin manifestations improved gradually.
Figure 1
A – Generalized, erythematous and poikilodermatous patches and thin plaques presented on the trunk and extremities. B – The lesions were typically atrophic, telangiectatic, and intermingled with mottled hyperpigmentation and hypopigmentation. C – Histopathology showed epidermotropism of atypical lymphocytes without spongiosis, vacuolar alterations of the basal layer, scattered melanophages, and lymphocytic infiltration in the papillary dermis (HE 100×)
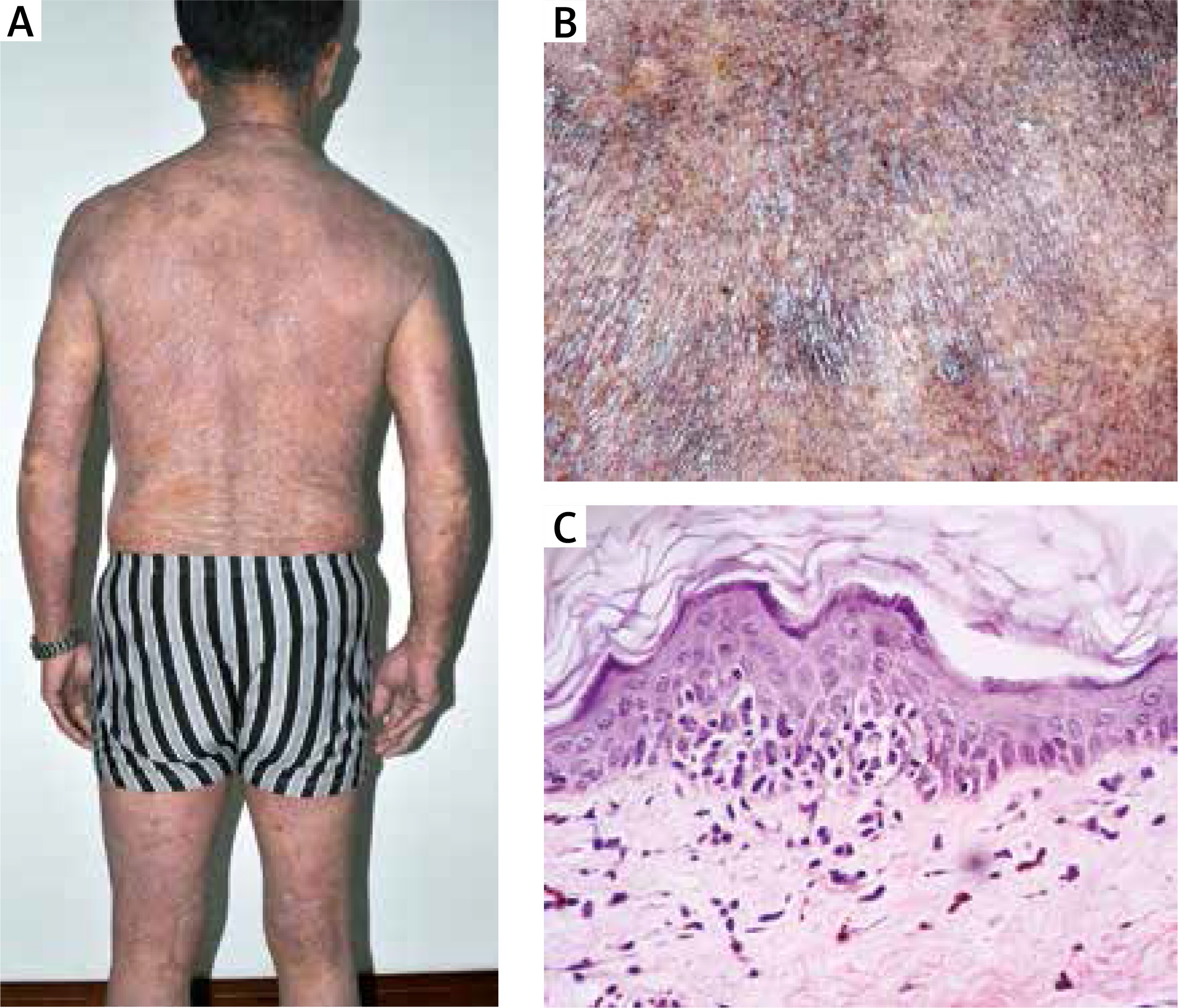
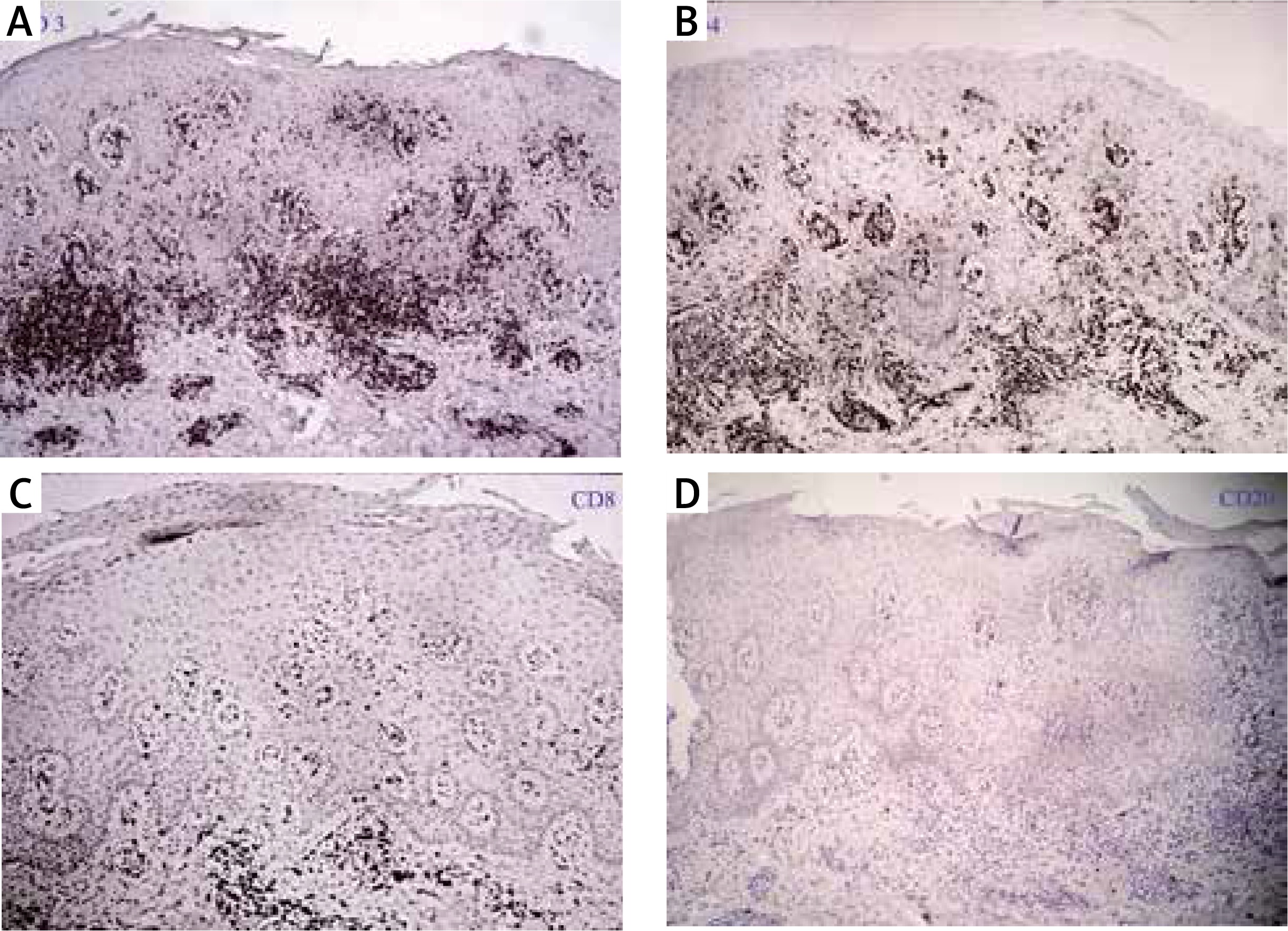
Figure 2
Immunohistochemically, the epidermotropic lymphocytes were positive for CD3 (A) and CD4 (B) while were negative for CD8 (C) and CD20 (D) (SP 100×)
Poikilodermatous MF, formerly referred to as poikiloderma vasculare atrophicans, is a variant of MF with an 11.2% incidence [2]. It has an earlier age of onset (44 years on average) in comparison with classic MF (55–60 years) [1]. The median duration of eruptions before diagnosis is 10 years in this variant compared with an average of 6 to 7 years for the classic MF. A male preponderance was shown by a male-to-female ratio of 1.6 to 2.0 [1].
Poikilodermatous MF usually presents with erythematous patches and plaques over non sun-exposed areas in the initial stage. Later, it develops to widespread or isolated poikiloderma with typical features of reticulate pigmentation, atrophy, scaling, and telangiectasia, which is predominantly located on the breast, abdomen, buttocks, and flexures [3]. These lesions may be asymptomatic or mildly pruritic.
Histopathology of poikilodermatous MF features an atypical T-cell infiltrate in the papillary dermis with signs of epidermotropism, epidermal atrophy and vacuolar changes at the dermal–epidermal junction [4]. Although Pautrier’s microabscesses are rarely present, lymphocytes in the epidermis are usually larger and more pleomorphic than those present in the dermis and often line up along the basilar layer as single cells or small clusters. Some cells exhibit perinuclear halos [5]. Keratinocyte apoptosis, which is not observed in classic MF, can also be observed. In the papillary dermis, prominent fibrosis may lead to a “wiry” collagen appearance. Scattered melanophages and telangiectasia are additional findings in the dermis. The immunohistochemical profile of atypical lymphocytes in poikilodermatous MF typically demonstrates a mature T-helper memory cell phenotype, which includes CD3+, CD4+, CD8-, CD30-, CD45RO+, and CD56-. Rarely, cases of poikilodermatous MF are characterized by a CD8+ cytotoxic T-cell phenotype, although it has a similar prognosis to the classic CD4+ CD8- phenotype [2, 6]. Gene rearrangement analysis in some patients may show the presence of monoclonal-type T cells.
Because widely-accepted criteria for poikilodermatous MF are still lacking, the diagnosis must be based on a combination of clinical demonstrations, histopathologic features, TCR gene analysis, and immunopathological criteria. This patient has a poikilodermatous lesion persisting for over 34 years. He was ultimately diagnosed with poikilodermatous MF (stage IB, TNMB classification for cutaneous T-cell lymphoma) and was placed at four points in the Algorithm for the Diagnosis of Early MF adapted from Pimpinelli [7]. Narrow-band ultraviolet B phototherapy is recommended as a safe and first-line therapy for treatment. If the patient fails, systemic therapies such as oral retinoids and a interferon are expected to be effective.
Poikilodermatous MF is indolent overall and has a better prognosis than classic MF. In a recent retrospective study, 96% of the study patients with poikilodermatous MF remained at the same stage over a mean follow-up period of more than 11 years [5]. Inasmuch as our patient had a 34-year duration of such a widespread disease, a stringent follow-up should be carried out to guarantee a better clinical outcome.