







Acute syndrome of apoptotic pan-epidermolysis (ASAP) is a recently described syndrome that is characterized by acute and massive cleavage of the epidermis resulting from hyperacute apoptotic epidermal basal cell injury [1]. This phenomenon is classically seen in drug-induced toxic epidermal necrolysis (TEN), but occasionally occurs in other conditions called ‘TEN-like’ diseases (e.g., lupus erythematosus (LE), acute graft versus host disease (GVHD) and pseudoporphyria) [2, 3].
Here we report a patient with the diagnosis of chronic cutaneous LE who progressed to SLE, developed ASAP while using nonsteroidal anti-inflammatory drugs and successfully treated with systemic prednisolone and intravenous immunoglobulin (IVIg) combination.
A 33-year-old female presented with 5-day history of painful and pruritic erythematous rash on her back, chest and face. In a couple of days lesions progressed to the hands, feet, and mucosal areas such as the mouth, eyes and vulva.
She carried the diagnosis of chronic discoid lupus erythematosus with disfiguring lesions located on the face and upper arms for 13 years. She had a history of spontaneous abortion at 16th week of gestation and had one healthy child.
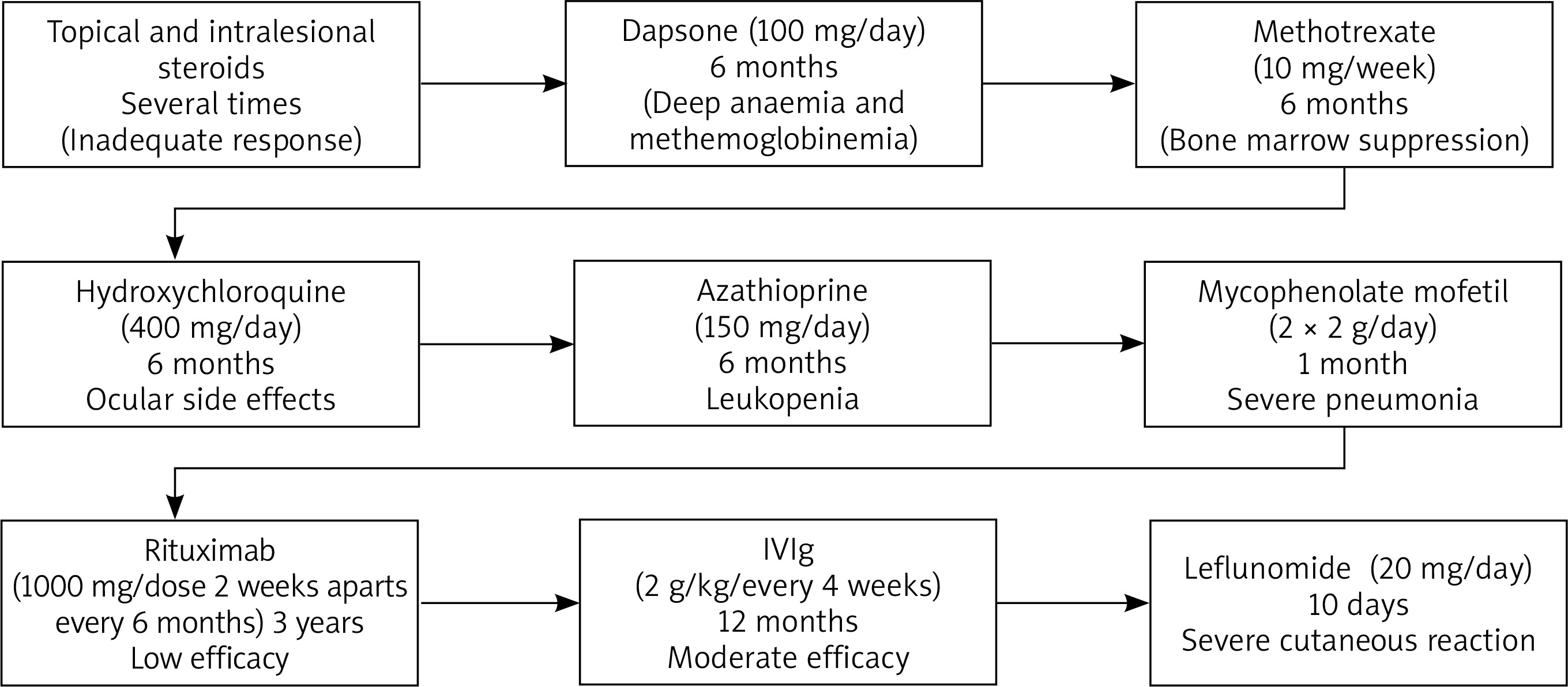
Since the onset of her disease she had been treated with topical and intralesional steroids, dapsone (100 mg/day, stopped because of deep anaemia and methemoglobinemia) methotrexate (10 mg/week, stopped for bone marrow suppression), hydroxychloroquine (400 mg/day, stopped due to ocular side effects), azathioprine (150 mg/day, discontinued after 6 months because of leukopenia), mycophenolate mofetil (2 × 2 g/day, stopped for severe pneumonia). Since she was intolerant to most of the standard immunosuppressives and lesions were in the active stage, rituximab was tried for 6 cycles (1000 mg/dose 2 weeks apart every 6 months) with no success, which led to initiation of IVIg 2 g/kg/month that was used for approximately 1 year. During this period, the patient was diagnosed with focal epilepsy that resolved with use of carbamazepine for 3 months.
One month prior to presentation to our clinic, she developed arthritis and arthralgia and her disease progressed to the systemic form. She was started on leflunomide (20 mg/day) but developed fever and erythematous rash on her face and upper trunk 10 days into treatment. The patient’s treatment course was summarised in Figure 1.
At presentation, the dermatologic examination showed macular exanthema and target-like lesions with bullae and erosions, oral mucositis, conjunctivitis and genital erosions (Figures 2 A, B). The laboratory examination revealed anaemia (9,5 mg/ml), elevated erythrocyte sedimentation rate (ESR) (22 mm/h), decreased C3 (0.723 g/l), C4 (0.0675 g/l), elevated ANA and anti-dsDNA titres.
Figure 2
A, B – Severe bullae and erosions on the face and trunk. C, D – Resolution of the lesions with postinflammatory hyperpigmentation after 6 months of therapy

The microscopic examination revealed subepidermal bulla formation caused by disseminated basal keratinocyte apoptosis and small pauci-inflammatory vesiculobullous tissue pattern.
Within the superficial dermis there was a mild mononuclear infiltrate with perivascular and periadnexal exocytosis of lymphocytes into the hair follicle with keratinocyte apoptosis and vacuolar degeneration (Figure 3 A, B).
Figure 3
Subepidermal bulla formation caused by disseminated basal keratinocyte apoptosis 100× (A), 200× (B), H + E

In addition to the cutaneous lesions, bilateral palpebral oedema, conjunctival erythema and erosions were noted. An ophthalmologic examination showed central corneal oedema with symblepharon formation associated with decreased vision in the left eye.
For systemic treatment, methylprednisolone of 80 mg/day was initiated and because of rapid progression, the dose was increased to 120 mg/day. On the fourth day of hospitalization, the patient’s rash became bullous and confluent, ultimately involving more than 50% of the body surface area, including the oral mucosa and lips, associated with elevation of liver function tests and decreased white blood cell count. She was then switched to 3 days of intravenous methylprednisolone and IVIg 3 g/kg for the next 3 days.
She was continued on a slow oral prednisolone taper. IVIg treatment was continued for 6 months at 2 g/kg/cycle. At 6-month follow-up, all her lesions resolved with postinflammatory hyperpigmentation (Figure 2 C, D). Corneal lesions resolved with central scarring and led to corneal transplantation.
Acute syndrome of apoptotic pan-epidermolysis (ASAP) is a relatively new entity. It is thought to be TEN that is induced by drugs in the course of lupus erythematosus. Classic TEN, other drug-induced eruptions and blistering skin disorders which may be found in lupus patients should be considered in differential diagnosis [2]. It must also be differentiated from a severe variant of Rowell’s syndrome which is characterized by recurrent erythema multiforme-like lesions in patients with all subtypes of lupus and suggested to be a limited form of ‘TEN-like’ acute or subacute cutaneous lupus erythematosus [4, 5]. Nevertheless, these patients usually have chilblains, speckled ANAs pattern, positive rheumatoid factor and anti-La/SSB antibodies; however, progression to TEN has not been noted [6]. It is very likely that Rowell’s syndrome and ASAP with TEN-like lesions may be part of the same spectrum of the illness [2, 7].
Patients with SLE are usually on multiple medications, which increase the risk of reactions like TEN, which is known to occur more commonly in patients with connective tissue disease [8]. It is still unclear whether the occurrence of ASAP in SLE patients is related to immune dysregulation or increased use of medications [9].
This patient with a history of DLE for a long period had progressed to the systemic form, presented with a widespread vesiculobullous eruption following the initiation of leflunomide, which is thought to be one of the triggers of this reaction.
The histopathological examination revealed epidermal detachment, which is suggestive of TEN-like eruption. Since TEN requires higher doses of IVIg, 3 mg/kg/cycle IVIg was given once and maintained at 2 mg/kg dose for 6 months with prednisolone.
There is a growing body of literature regarding the use of IVIg with a combination of systemic steroids and wound care in TEN-like eruptions in SLE but large prospective studies are still lacking [8, 9]. Most of the reported cases survived and entered remission.
In conclusion, ASAP usually occurs in the drug-induced setting in the course of lupus. Because of the complexity of the underlying disease itself and the inherent multiple medication use in lupus patients, clinicians need to be aware of complex presentation and rapid progression of bullous lesions in patients with SLE.