





Hypoxia affects the majority of critically ill patients and continues to be a common cause of morbidity and mortality [1, 2]. During hypoxia, oxyge-na-tion may fall below the level necessary to sustain life [3]. The lowest documented PaO2, recorded near the summit of Mount Everest in a climber breathing ambient air, was 19.1 mm Hg [3]. This was documented during the Caudwell Extreme Everest Expedition in 2007, a study of 222 climbers exposed to extreme altitudes above 5500 metres [3]. From this and other expeditions to Mount Everest, we know that humans exposed to extreme altitudes can survive through physiological acclimatization responses, primarily hyperventilation and resulting hypocapnia [4, 5]. To our knowledge, many of these findings have not been applied to clinical practice in the acute management of hypoxia.
Under profound hypoxic conditions, when intracellular O2 decreases, the expression of hypoxia-inducible factor-1α (HIF-1) takes place within 2 minutes [2, 6]. HIF-1, through tyrosine hydroxylase, increases the carotid body sensitivity, which plays a crucial role in the acclimatization response to hypo-xia by inducing tachypnoea [7]. This increases alveolar ventilation, generating hypocapnia to avert a drop in alveolar partial pressure of O2 (PAO2), which could fall to levels incompatible with life [2]. Additionally, the HIF-1 promotes the expression of vascular endothelial growth factor, which promotes angiogenesis and neural plasticity of the respiratory centre. Both mechanisms play a key role in chronic hypoxia [7, 8].
A common concern regarding hyperventilation is the potential reduction in cerebral blood flow due to hypocapnia-induced cerebral vasoconstriction. This has been measured in normoxic patients using transcranial doppler and positron emission tomography [9, 10]. Under hypoxic conditions the opposite is true, because different mechanisms, including adenosine, substance P, nitric oxide, and prostaglandins, mediate cerebral vasodilation [11]. This hypoxia-induced vasodilation opposes hypocapnia-associated vasoconstriction and increases cerebral blood flow [12]. One study demonstrated cerebral blood flow approximately 5 times higher at a PaO2 of less than 25 mm Hg <i>versus</i> a normoxic PaO2 [2]. Furthermore, at high altitudes, climbers with PaCO2 levels as low as 7–8 mm Hg and averaging 13.3 mm Hg have shown unimpaired cognition, confirming sufficient cerebral blood flow [3, 4]. These results shed light on the possibility of implementing new rescue manoeuvres for the management of acute severely hypoxaemic patients. However, there are concerns regarding neuronal excitation triggered by severe hypoxic-hypoxia and tachypnoea. Seizures have been reported in climbers, especially in those with a previous history of seizures [2, 13].
We hypothesized that mimicking the physiological hyperventilation necessary to survive extreme hypoxic conditions, such as those observed at high altitudes, minimizes the haemodynamic and serum chemistry derangements caused by profound acute hypoxia. To test this hypothesis, our primary aim was to evaluate the impact of artificially induced hyperventilation on haemodynamic status, serum lactate and glucose levels, and arterial blood gases in a deeply anaesthetized terminal rat model of profound acute hypoxic hypoxia.
METHODS
Experimental design
After receiving approval by the Institutional Animal Care and Use Committee of the University of Texas MD Anderson Cancer Center, we conducted a terminal rat model of acute hypoxic hypoxia using 24 male Sprague-Dawley spontaneously breathing rats (329–419 g) aged 8–12 weeks. The rodents were deeply anaesthetized and mechanically ventilated for 3 hours; no muscle relaxants were used in any of the study groups. Additional analgesics were unnecessary because the animals were under total intravenous anaesthesia using pentobarbital. All the rats were euthanized with pentobarbital according to institutional protocols at the termination of the experiment. We administered equal ventilation to all the study animals with a target PaCO2 of 40 mm Hg and an FiO2 of 1 for the first hour of the experiment, to achieve steady-state conditions. For the rest of the experiment, the rats were randomly allocated to 3 different groups. The control group comprised 5 rats mechanically ventilated at FiO2 = 1.0. The hypo-xic spontaneously-hyperventilating (HSH) group comprised 10 rats that were mechanically ventilated at FiO2 = 0.08, and the hypoxic artificially induced hyperventilation (HAIH) group comprised 9 rats that were mechanically ventilated at an FiO2 = 0.08 with a respiratory rate targeting a PaCO2 = 10 mm Hg.
FiO2 = 0.08 was chosen for the HSH and HAIH groups to mimic a partial pressure of inspired oxygen (PIO2) of 57 mm Hg, similar to that encountered at an altitude of approximately 7100 m. In previous studies of high-altitude medicine, the PaCO2 level ranged between single digit mm Hg to the low teens; therefore, a level of 10 mm Hg was selected as a target for our experiment [3, 14]. The target PaCO2 was achieved in the HAIH group by hyperventilating the animals to mimic the acclimatization response.
Instrumentation
Animals were cared for and managed under strict Institutional Animal Care and Use Committee standards throughout the study. On the day of the experiment, animals were taken to the anaesthesia room and weighed using a microbalance. Each rat was injected with pentobarbital (50 mg kg-1 intra-peritoneally) in preparation for the procedure. Anterior neck and right groin incision sites were shaved using electric clippers to avoid skin trauma. The rats were then transported to the procedure room inside a covered animal cage. In the procedure room, after adequate anaesthesia was verified, they were placed over a warming pad set at a temperature of 36°C. A rectal temperature probe was secured in place, and the temperature was adjusted using both heating lamps and a heating pad to keep the rectal temperature at 36°C. Animals were adequately ventilated and monitored for a period of approximately 20 minutes. During this period, we monitored the haemodynamic state of the animals through skin colour, respiratory rate, and end tidal CO2 (ETCO2). The animals were expected to achieve a deep enough level of anaesthesia to proceed with the experiment without causing them harm or undue stress. Before skin incisions were performed, 2 mL of Plasma-Lyte (Baxter International Inc., Deerfield, USA) solution, a balanced salt solution with a pH of 7.4, was injected intravenously via the tail vein via 21-gauge needle to ensure adequate hydration. Once the bolus was injected, pressure was held over the venipuncture site to ensure haemostasis. Plasma-Lyte solution was infused at a set rate throughout the experiment to avoid intravascular volume depletion.
Tracheostomies were performed using both sharp and blunt dissection via a 1-cm horizontal neck skin incision. The trachea was surrounded with two 2-0 silk sutures to gain adequate control of the airway during the tracheal incision. Immediately prior to incision, the ventilator (Harvard Apparatus, Holliston, USA) was turned on and set to the correct tidal volume and respiratory rate according to the animal’s weight. The oxygen flowmeter connected to the ventilator was set at 2 L min-1. The trachea was then incised. A 16-gauge IV catheter Angiocath (Becton, Dickinson and Company, Franklin Lakes, USA) was inserted into the tracheal lumen and advanced proximally. The Angiocath was secured to avoid tube migration into the right or left main bronchus. The Angiocath, working as an endotracheal tube, was connected to the ventilator breathing circuit. ETCO2 was immediately verified using a 5440 Anesthesia Monitor Capnograph (Ohio Medical, Gurnee, USA). The average peak pressure was also monitored to ensure the correct placement of the tracheostomy tube. An oximeter was used to verify that the correct FiO2 was delivered. Once oxygenation and ventilation were verified, a 2 cm right groin incision was performed to gain access to the right femoral vessels. Central venous access and arterial line access were obtained via the right femoral vessels using appropriate size catheters with minimal blood loss. The central venous line was connected to the pump which was pre-programmed to infuse pentobarbital at 20 mg kg-1 h-1 intravenously to establish maintenance total intravenous anaesthesia. The arterial line and its pressure transducer were connected to a Propaq 202 EL Encore anaesthesia monitor (Welch Allyn, Skaneateles, USA). The arterial waveform was verified, and the arterial line was zeroed in preparation for the acquisition of baseline haemodynamic readings. Systolic, diastolic, and mean arterial blood pressures were recorded. The arterial line allowed baseline as well as maintenance blood sampling.
Blood loss was measured by weighing all 4 × 4 gauze pads employed to recover any blood extravasated during the surgical procedure. All blood samples obtained for chemistry determinations were also accounted for in our process. A baseline haemoglobin level was obtained at the beginning of the procedure as well as during the experiment to rule out anaemia, which if present would affect the data analysis. Individual blood samples for analysis, obtained via the femoral arterial line, amounted to 0.2 mL each. An iSTAT (Abbott Laboratories, Chicago, USA) point-of-care device was employed for all chemistry analyses.
Measurements
Laboratory measurements, including arterial oxygen saturation (SaO2), PaO2, PaCO2, arterial O2 content (CaO2), arterial pH, and serum levels of lactate and glucose, were recorded at 60-, 120-, and 180-minute time points. The mean arterial pressure (MAP) was recorded at 5-minute intervals throughout the intervention.
Statistical analysis
Sample sizes were estimated based on previous similar studies. Blinding methods were not used when assessing results. Alterations in haemodynamics and serum blood chemistry were examined, with the rat as the unit of analysis. Summary statistics such as the number of missing observations, mean, median, standard deviation, minimum, and maximum were determined for continuous data. The Wilcoxon rank-sum test was used to compare the distributions of measurements between the HSH and HAIH groups. The Kruskal-Wallis test was used to compare the distributions of measurements among the control, HSH, and HAIH groups. Mean and standard error bar graphs over time points for each metric were generated by the treatment group. All tests were 2-sided, and P-values less than 0.01 were considered statistically significant. Outliers were not evaluated, and no data were excluded from statistical analysis. All analyses were conducted using SAS (v9.1, Cary, North Carolina, USA) and GraphPad Prism 8 (V8, La Jolla, California, USA).
RESULTS
During the experiment, 2 rats (2/10) in the HSH group died within the first hour of receiving oxygen at FiO2 = 0.08, and their data was excluded from data analysis (Figure 1A). One rat (1/9) in the HAIH group did not complete the study due to technical issues with the calibration of the FiO2, and its data was excluded from data analysis.
FIGURE 1
Artificially induced hyperventilation in hypoxic rats. A) Flowchart of the studied groups. Studied rats were deeply anaesthetized and mechanically ventilated for 3 hours. No muscle relaxants were used in any of the study groups. During the first hour of the experiment, all the rats were ventilated at an FiO2 of 1 to achieve steady state. Then the rats were randomly allocated to 3 different groups. The control group (n = 5, green) that was mechanically ventilated at an FiO2 of 1, the hypoxic spontaneously hyperventilating group (HSH, n = 10, blue) that was mechanically ventilated at an FiO2 of 0.08, and the hypoxic artificially induced hyperventilation group (HAIH, n = 9, red) that was mechanically ventilated at an FiO2 of 0.08 with a respiratory rate targeting a PaCO2 of 10 mm Hg. B) Mean arterial pressure (MAP) in the studied groups at different time points. MAP for each group is presented at 10-minute intervals throughout the 3-hour experiment period. The data is represented as mean, and the standard error is shown at 120 and 180 minutes. Differences among the HAIH and HSH groups were calculated with the Wilcoxon rank-sum test. Statistical significance was accepted as P < 0.01. During the first hour of the study there were no differences in MAP among the 3 groups. At the end of the steady-state period, the MAP of the HSH and HAIH groups rapidly fell. However, after the acclimatization process driven by the hyperventilation, the HAIH group increased the MAP. At 120 and 180 minutes, the HAIH group had a significantly higher MAP (P < 0.005) than the HSH group
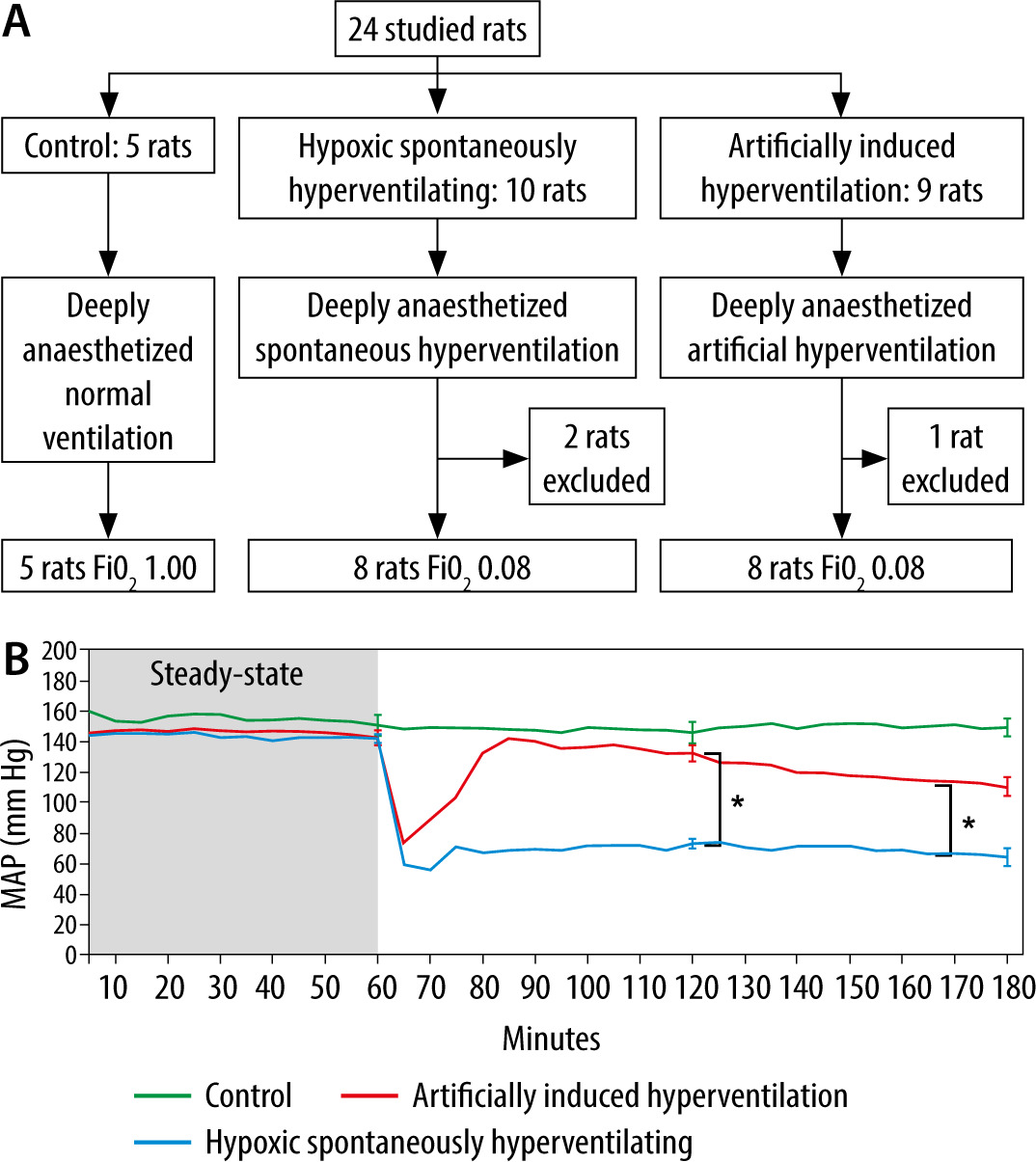
The HAIH group maintained significantly higher blood pressure levels compared to the HSH group, which suffered severe deterioration of their haemodynamic status (Figure 1B). At 120 minutes and 180 minutes, the HAIH group had a significantly higher MAP (P = 0.0048) than the HSH group. Animals in the HAIH group had significantly higher levels of SaO2 and CaO2 at 120 minutes and 180 minutes (P = 0.005), of PaO2 at 120 minutes (P = 0.007), and of glucose at 180 minutes (P = 0.007) than animals in the HSH group (Figure 2A and Figure 3A–C). Furthermore, the mean pH of the HAIH group was higher than those of the control and HSH groups at 120 and 180 minutes (P = 0.005) (Figure 2B). Animals in the HAIH group had significantly lower PaCO2 (P = 0.005, P = 0.005) and lower lactate (P = 0.021, P = 0.039) measurements at 120 minutes and 180 minutes than animals in the HSH group (Figure 2C and Figure 3D). Means, medians, standard deviations, and ranges of all laboratories performed are presented in Table 1.
FIGURE 2
Arterial blood gas results in rat groups at different time points. Arterial blood gases were measured at 60 minutes, 120 minutes, and 180 minutes during the experimental period in all rats. The data is presented in box-and-whisker plots. Whiskers depict the minimum and maximum values. The green boxes represent control rats, which were consistently ventilated throughout the experiment at an FiO2 of 1. The red boxes represent the hypoxic artificially induced hyperventilation (HAIH) group, which were ventilated during the first 60 minutes at an FiO2 of 1 and after that at an FiO2 of 0.08, with a respiratory rate titrated targeting a PaCO2 of 10 mm Hg. The blue boxes represent the hypoxic spontaneously hyperventilating group (HSH) group, which were ventilated during the first 60 minutes at an FiO2 of 1 and after that at an FiO2 of 0.08 without modification in respiratory rate. Differences among the HAIH and HSH groups were calculated with the Wilcoxon rank-sum test. Statistical significance was accepted as P < 0.01. A) The partial pressure of arterial oxygen was significantly higher for the HAIH at 120 minutes and had a tendency towards significance at 180 minutes (P = 0.013). B) The pH was significantly higher in the HAIH rats at 120 and 180 minutes (P = 0.005, P = 0.005). C) The partial pressure of arterial carbon dioxide was lower in the HAIH group (P = 0.005, P = 0.005)

FIGURE 3
Selected laboratory results in rat groups at different time points. Laboratory data were measured at 60, 120, and 180 minutes. The data are presented in box-and-whisker plots. Whiskers depict the minimum and maximum values. The green boxes represent the values of the control rats, which were consistently ventilated throughout the experiment at an FiO2 of 1. The red boxes represent the values of the hypoxic artificially induced hyperventilation (HAIH) rats, which were ventilated during the first 60 minutes at an FiO2 of 1 and then at an FiO2 of 0.08, with a respiratory rate titrated targeting a PaCO2 of 10 mm Hg. The blue boxes represent the values of the hypoxic spontaneously hyperventilating (HSH) rats, which were ventilated during the first 60 minutes at an FiO2 of 1 and after that at an FiO2 of 0.08 with no modification in respiratory rate. Differences among the HAIH and HSH groups were calculated with the Wilcoxon rank-sum test. Statistical significance was accepted as P < 0.01. A) Arterial oxygen saturation (SaO2) was significantly higher in the HAIH group at 120 and 180 minutes (P = 0.005, P = 0.005). B) Arterial oxygen concentration (CaO2) was significantly higher in the HAIH group at 120 and 180 minutes (P = 0.005, P = 0.005). C) Serum glucose levels (mg dL-1) was significantly higher in the HAIH group at 180 minutes (P = 0.007). d) Serum lactate was lower in the HAIH group at 120 and 180 minutes, but not statistically significant (P = 0.021, P = 0.039)
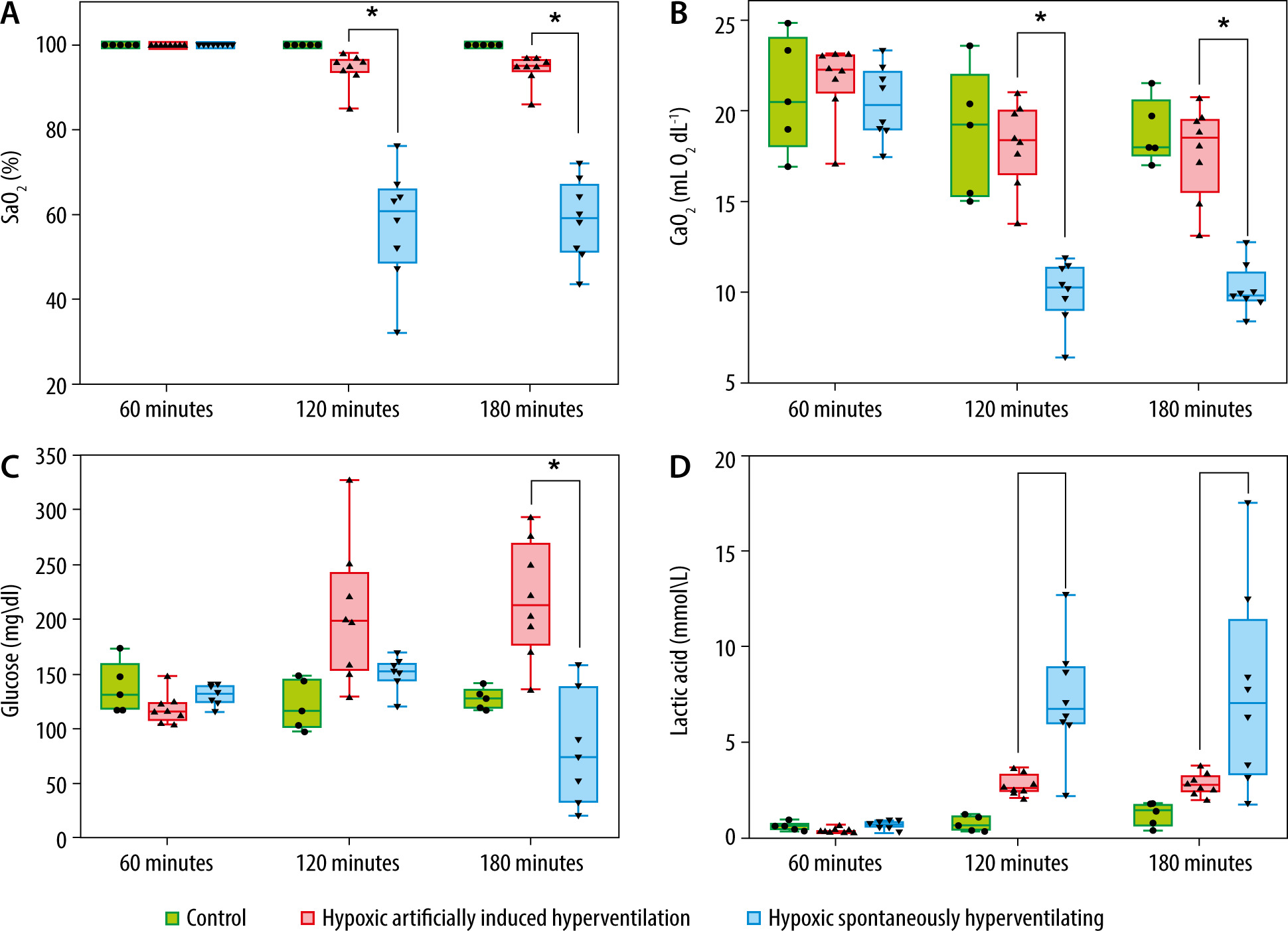
TABLE 1
Laboratory results by group and study time
Regarding ventilator parameters, the average peak pressure, tidal volume, respiratory rate, and minute ventilation were higher in the HAIH group of rats than in the HSH group at 120 and 180 minutes (P = 0.011) (Table 2).
TABLE 2
Ventilation parameters
[i] C – control group, HAIH – hypoxic artificially induced hyperventilation group, HSH – hypoxic spontaneously hyperventilating group. *Minute ventilation in the HSH group is not accurate given that it only takes into consideration the ventilator settings. Rats in this group were not paralyzed and therefore were left to regulate their own respiratory rate
DISCUSSION
The results of this study confirm our hypothesis that artificially induced hyperventilation significantly reduced the negative impact of hypoxic-hypoxia on haemodynamic status and serum chemistry parameters. Additionally, the HAIH group demonstrated adequate oxygenation and proper aerobic metabolism leading to normal serum glucose and lower serum lactate levels during the period of extreme hypoxia as compared to the HSH group. Despite the higher peak airway pressure of the HAIH rats, which had a tendency towards statistical significance compared to the HSH group, we observed an increased MAP accompanied by an improved biochemical profile, possibly due to the more adequate oxygenation. The HAIH group exposed to 8% oxygen had a CaO2 similar to that of the control group exposed to 100% oxygen (Figure 3B). The HAIH and HSH study groups displayed a MAP decrease after the initiation of the experiment (Figure 1B). This circulatory crisis is probably due to a state of heart failure secondary to the depressant effect of hypoxia on the myocardium [15]. If these findings are successfully translated to the clinical setting, they may change the way we manage acute, severe hypoxaemia.
To understand how hyperventilation protects against hypoxia through acclimatization, both respiratory and metabolic mechanisms must be considered. These have been studied extensively in the field of high-altitude medicine and physiology [16]. At high altitudes, the most important factor of pulmonary gas exchange is the increased alveolar ventilation, which is inversely proportional to PaCO2 [14]. As barometric pressure decreases, the PaO2 is reduced. Alveolar partial pressure of CO2 (PACO2) within a normal range would further decrease the PaO2. The PAO2 level could be dangerously low and cause a situation that would be incompatible with life [2]. Consequently, tachypnoea sustained by climbers exposed to extreme conditions can dramatically reduce their PaCO2 and allow them to survive at high altitudes with PIO2 as low as 43.1 mm Hg, representing less than a third of the PIO2 observed at sea level [3]. In an effort to imitate climbers’ survival mechanisms under hypoxic conditions in the artificially induced hyperventilation group, we induced hyperventilation targeting a PaCO2 of 10 mm Hg to mimic high-altitude acclimatization.
The impact of hypocapnia on the oxygen affinity of haemoglobin must be considered, because increased oxygen affinity caused by low PaCO2 can be ascribed to the increase in the pH [2]. Earlier studies examining the oxygen dissociation curve in the presence of limited oxygen diffusion across the blood-gas barrier found that a leftward shift of the curve resulted in a higher pressure of mixed venous blood oxygen, an index of tissue oxygenation [2, 17]. This suggests that in the presence of limited oxygen diffusion, increased haemoglobin affinity enhancing oxygen loading in the alveoli outweighs the negative impact on oxygen unloading in tissues [17–19]. At extreme altitudes, hyperventilation allows climbers to increase their oxygen affinity through respiratory alkalosis, reaching a CaO2 adequate for tissue oxygenation [20]. This observation was replicated in the HAIH group, which sustained a very low mean PaO2, while its mean CaO2 approximated that of the control group.
Although the concern for cerebral vasoconstriction secondary to hypocapnia is valid in normoxic conditions, studies have shown that this risk does not apply to hypoxia [9, 10]. In hypoxia, the vasoconstriction generated by hyperventilation is counteracted by compensatory arterial vasodilation [11, 12], allowing adequate brain oxygenation. Moreover, it has been documented that at altitude, global cerebral blood flow increases through acclimatization to maintain cerebral oxygen delivery [21, 22].
In order to achieve acclimatization by hyperventilation, respiratory muscle integrity is essential. The striated muscle of the respiratory apparatus has the capacity to sustain considerable tachypnoea. The extraordinary capacity of this muscle to avoid fatigue is driven by its oxygen-conforming nature, which allows the matching of oxygen consumption (VO2) to oxygen delivery (DO2) [23, 24], but for this to take place a constant source of energy must be secured.
During hypoxia, metabolic changes activate anaerobic pathways. In anaerobiosis, glycogen is quickly depleted, and serum lactate levels rise steeply [25]. Lactate becomes an essential source of energy in hypoxic-hypoxia and is utilized directly by a variety of tissues [24, 26]. In the heart, lactate is consumed via anaplerosis, replenishing intermediaries of the Krebs cycle, and has even been shown to be haemodynamically protective [27–29]. How-ever, if the striated muscle is fatigued and not able to compensate for the oxygen depletion, the endpoint will be severe deterioration of the haemodynamic status, as observed in the HSH rats. The HAIH group withstood the hypoxic conditions while maintaining adequate MAP and tissue oxygenation, indirectly shown by signs of aerobic metabolism, with a lower mean lactate level (2.8 mmol L-1) and high levels of glucose at 180 minutes (Figure 3).
Lastly, we observed that the PaCO2 in the HSH group of rats also decreased in comparison with the control group. This was expected because no muscle relaxants were used, allowing them to put into motion their physiological compensatory mechanisms as a survival response. This brought increased work of breathing and increased serum lactate. While lactate was increased compared to the control group, the pH was similar, which may be explained by respiratory alkalosis responding to an acute effect caused by the washout of PaCO2 by hyperventilation. During acute hypoxic-hypoxia, the physiological response in the HSH rats differed from that in HAIH. HSH rats demonstrated a decrease in PaCO2 product of normal compensatory tachypnoea. Nevertheless, the pH remained in the normal range, possibly due to the elevated lactic acid. In contrast, the induction of HAIH significantly raised the serum median pH to 7.88, with the associated haemodynamic and biochemical beneficial responses seen during acclimatization.
Our study had a number of limitations. First, we were unable to determine the amount of time HAIH can be sustained without causing deleterious effects, as well as the severe complications derived from the prolonged exposure to severe alkalosis, particularly within the central nervous system. Second, we did not measure cardiac output and central venous saturation (SvO2), which could provide useful information about DO2/VO2 ranges. Third, the animals’ respiratory rate and minute ventilation of the HSH group are not accurate, given that the respiratory rate reported was obtained from the ventilator and did not include the spontaneous breaths of the hypoxic animals, because the tachypnoea generated by hypoxia made it challenging to monitor their true respiratory rate. This is a crucial clarification, because no muscle relaxants were used on any of the study animals, and the observed hyperventilation on the non-hyperventilated rats was secondary to the acclimatization response to hypoxia. Lastly, we used a hyperoxic control group of animals ventilated with an FiO2 of 1. Investigators aiming to replicate this study should consider adding an additional group of normoxic animals that could exclude the effect of hyperoxia.
CONCLUSIONS
Artificially induced hyperventilation using mechanical ventilation in an acute hypoxic-hypoxia animal model led to the correction of severe serum chemistry derangements, haemodynamic stability, and arterial oxygen content. This may represent a novel rescue manoeuvre during acute hypoxic-hypoxia by stabilizing a rapidly deteriorating situation buying time we may not otherwise have, and serve as a bridge to a more permanent form of support. These experimental findings should be further studied and validated before being translated to the clinical setting.