







Blueberry muffin baby syndrome is a rare and non-specific clinical presentation in newborns, characterized by the presence of widespread, maculopapular lesions of blue-red or violaceous colour and cohesive consistency [1]. The term “blueberry muffin baby” was initially coined to describe the skin manifestations of congenital rubella during the American epidemic in the 1960s [2, 3]. The presence of skin lesions is secondary to extramedullary hematopoiesis, which can result from intrauterine viral infections (TORCH syndrome), hematologic dyscrasias (twin-to-twin transfusion syndrome, hereditary spherocytosis, haemolytic disease of the newborn) or neoplasms (mastocytosis, histiocytosis, neuroblastoma, rhabdomyosarcoma, leukaemia) [1, 3].
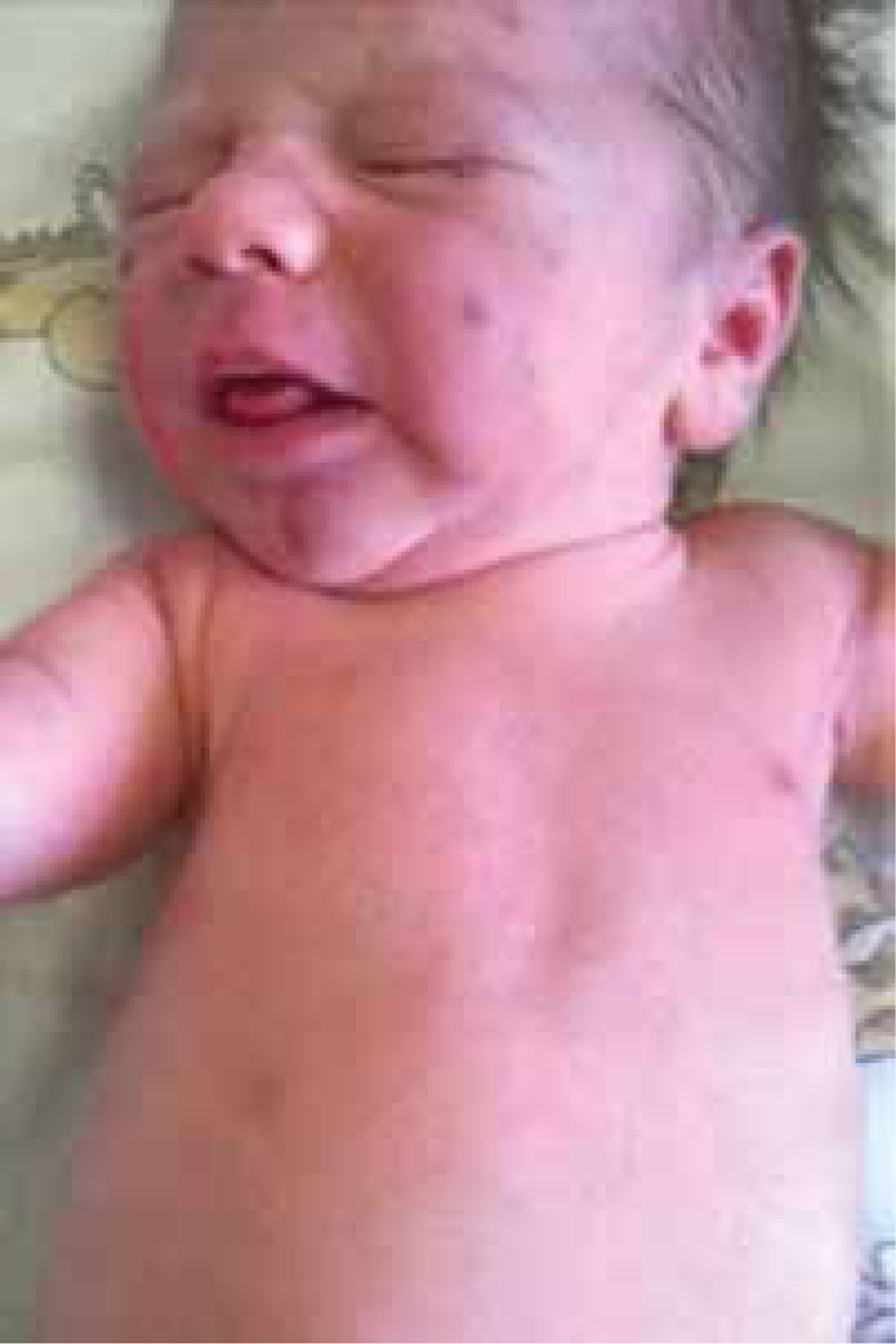
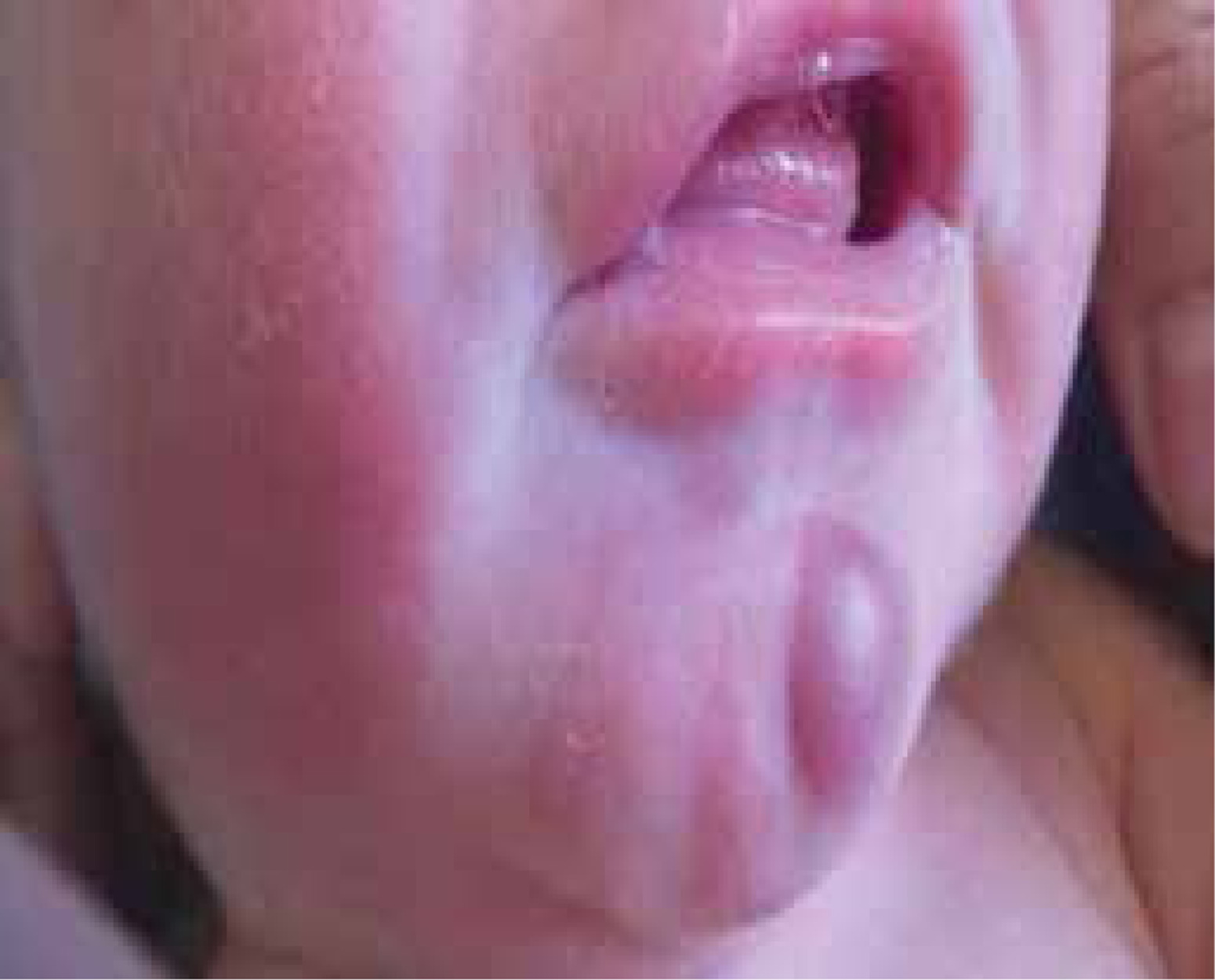
We present the case of a male infant with a birth weight of 3320 g, Apgar score 10/10, blood type AB RhD +, who was born in gestation week 39 by normal vaginal delivery. The mother was a healthy 33-year-old woman and the father was a 32-year-old man, without any specific family history. The early stages of pregnancy had been complicated by urinary infection, successfully treated with oral antibiotics. HIV and very VDRL were non-reactive. On skin examination it was noticed that the baby had a “blueberry muffin appearance” with widespread purple papules and nodules, up to 1 cm in diameter, disseminated on the scalp, face, truck and limbs (Figures 1 and 2). The lesions deteriorated in the following days. The results of physical examination were otherwise unremarkable.
Laboratory tests showed a decrease in white blood cells (WBC values ranged from 6.6 to 7.2 × 109) and the differential blood count (DBC) revealed neutrophils 11%, eosinophils 9%, and lymphocytes 74%. A prolonged prothrombin time (PT up to 16.78) and international normalized ratio (INR) (up to 1.23), and mild elevation of aspartate aminotransferase and hyperglycaemia were noted. Haemoglobin, haematocrit, platelet count, electrolytes and urine analysis were normal for the age. A transient increase in C-reactive protein (up to 38.82 mg/dl) was also observed. Repeated blood cultures were negative. Serology testing excluded congenital infection with toxoplasma, rubella, and cytomegalovirus. Abdominal, cardiac and cranial ultrasound scans showed no abnormalities. Ophthalmic examination showed oval optic nerve shield with peripheral pallor.
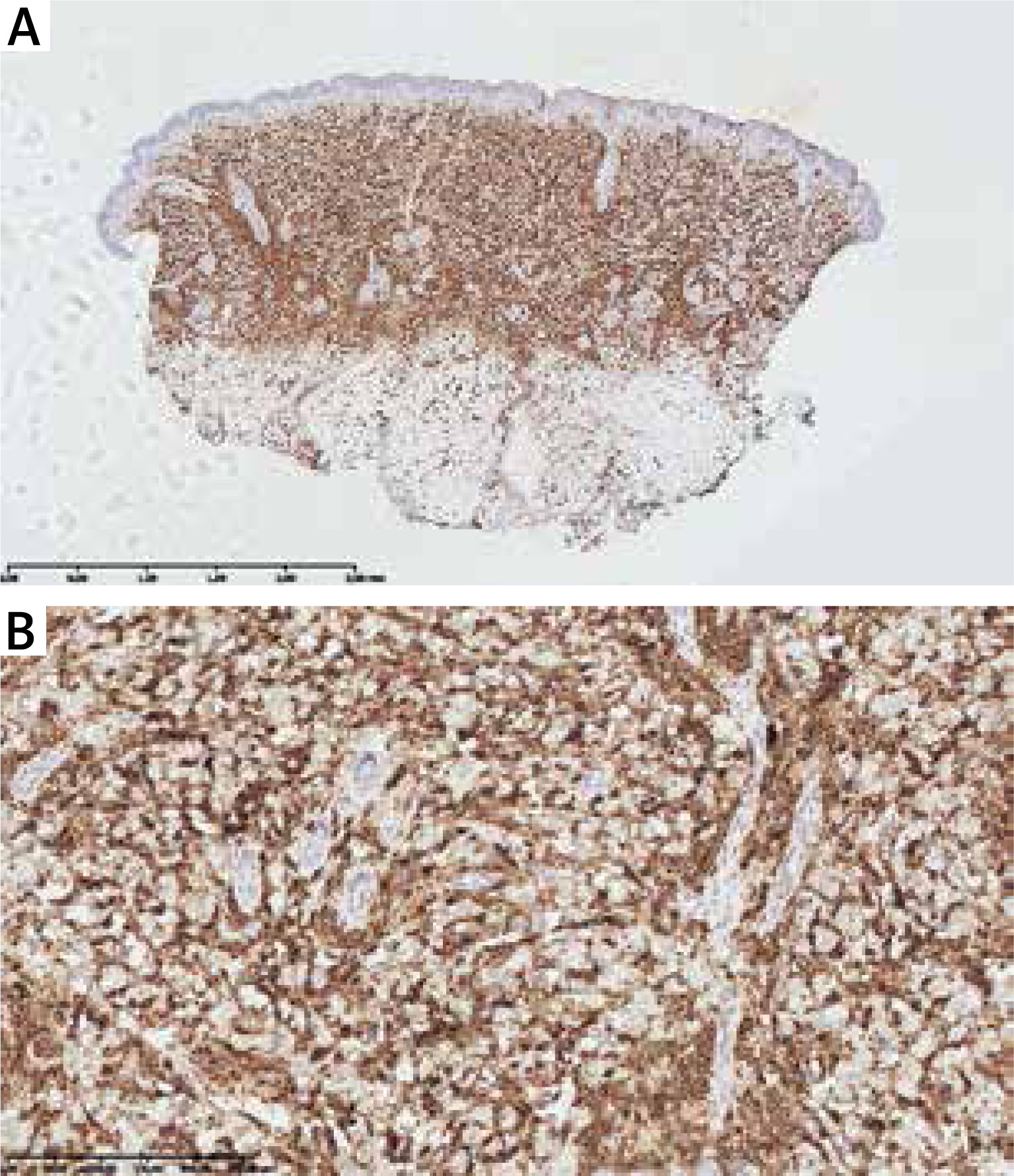
On the 2nd day of hospitalization the newborn revealed tachypnoea. Chest X-ray demonstrated symmetric linear opacities in upper pulmonary fields, and on transthoracic ultrasound a transient neonatal tachypnoea was confirmed. Systemic administration of amikacin and ampicillin was implemented, resulting in rapid improvement. Apart from the transient tachypnoea, the overall clinical condition was good, and at the same time skin lesions gradually progressed. On the 9th day the infant was referred to skin biopsy at the Haematology Department. Leukopenia with neutropenia (323/mm3) and lymphocytosis persisted. Histological examination of the suspicious nodule demonstrated a dense infiltrate of blast cells (Figure 3). Immunophenotyping showed a population of mononuclears expressing CD15, MPO, CD34, CD56, CD4, CD31, CD71, CD68/PGM, CD68/KP1 (Figures 4 A, B). Ki-67 proliferative index was 60%. Genetic testing towards Kostmann disease and inherited neutropenia turned out negative. No pathologic cells in cerebrospinal fluid were detected. Blastosis in myelogram was evaluated at 0% with granulocyte inhibition of maturation at the stages of myelocyte and promyelocyte.
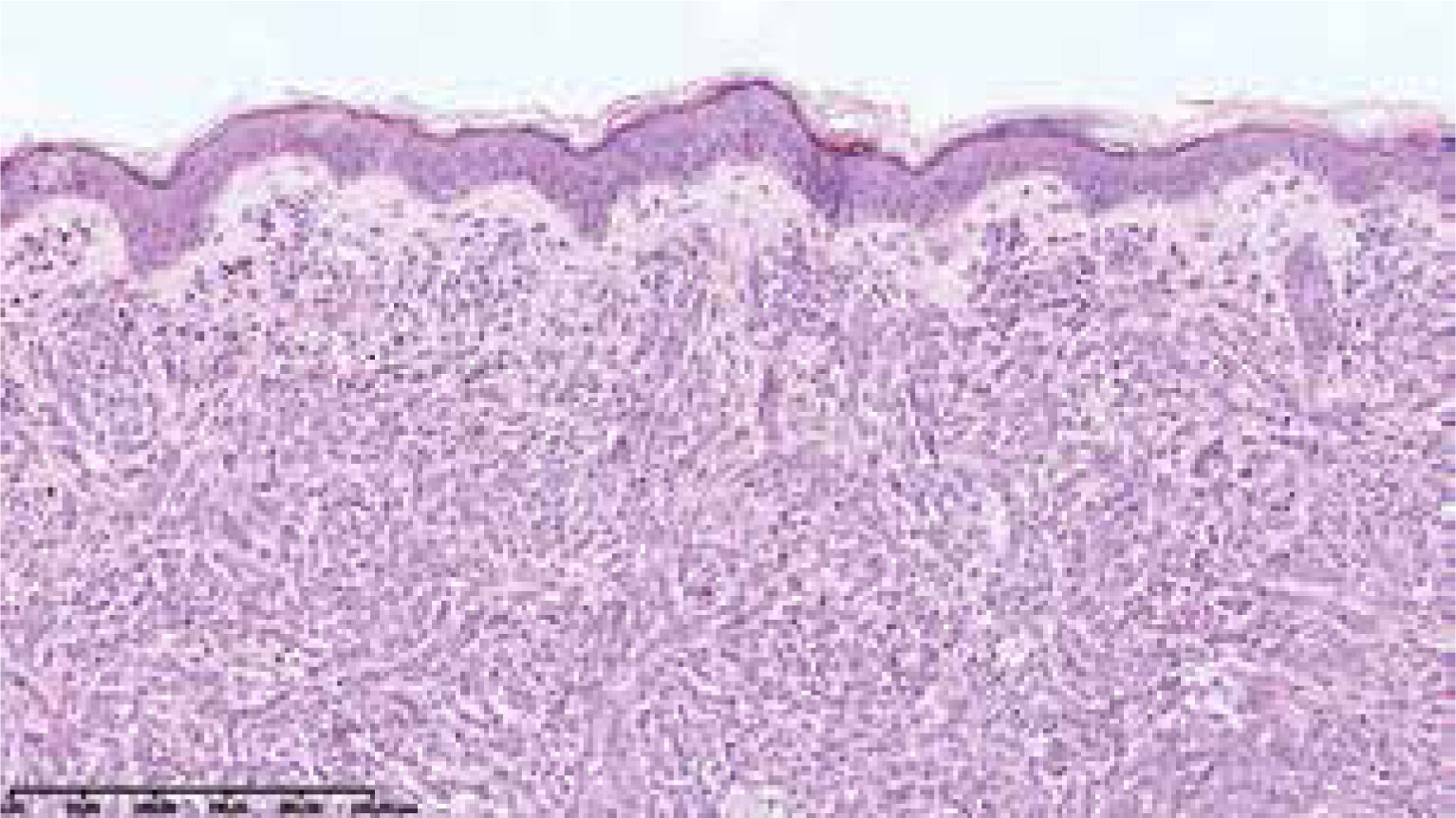
Figure 4
Dense infiltrates of mononuclears in dermis and subcutis with a positive CD15 immunohistochemistry staining (distinguishing marker for human myeloid cells; A – 2× magnification, B – 40× magnification, respectively)
A diagnosis of acute myeloid leukaemia, French-American-British (FAB) type M4, was made. The AML-BFM 2012 [4] treatment scheme was applied (AIE, Ara-C, HAM, AI/2-CDA). The therapy resulted in multiple septic complications and hematologic toxicity grade IV according to WHO. The remission was achieved, however after 6 months a relapse was confirmed in bone marrow and eye orbit histopathologic examination (cellular infiltrate positive for CD68+, CD 15+; Ki-67 was 70%). The patient’s disease progressed and he died within 12 months of diagnosis.
Congenital leukaemia (CL) is a term applied to leukaemia diagnosed within the first month of life [5–9]. The criteria for diagnosis are: (i) immature cell proliferation in myeloid, lymphoid, or erythroid series, (ii) infiltration into extrahematopoietic tissue, (iii) and absence of other disease mimicking this proliferation [5, 10, 11].
CL is an extremely rare entity with an estimated incidence between 4.3 and 8.6 per million live births and it accounts for 0.5–1% of all cases of childhood leukaemia with approximately 200 documented cases [5, 7, 9–13]. The majority of CL cases are of myelogenous type, with most common variants being myelomonocytic (M4) and monocytic (M5) FAB subtypes [5, 7, 10].
Symptoms suggesting leukaemia are: hepatosplenomegaly (present in 80% of cases), central nervous system involvement (50%), lymphadenopathy (25%), lethargy, pallor, anaemia and leucocytosis [10]. In the presented case those were lacking, making the diagnosis even more challenging. The hallmark feature in our patient was cutaneous infiltration by malignant leukocytes. The prevalence of leukaemia cutis ranges from 15% to 60% of infants with CL and it may be the first manifestation of systemic disease in a half of them [5, 7, 9, 14, 15]. Cutaneous infiltrations present most commonly as firm, freely movable violaceous nodules up to 1–2.5 cm in diameter, in generalized distribution, causing the blueberry appearance. Tumours, papules, plaques, macules and ulcers might also occur [6].
In histopathology the most common pattern is a dense infiltrate of leukemic cells in a linear pattern between collagen bundles in the reticular dermis, extending into the subcutis. Malignant myeloid cells are dominant in the infiltrate and they appear monotonous and homogenous cytologically [12].
The causes of CL are not elucidated, but the disease has been associated with maternal exposure to radiation, bioflavonoids in diet, marijuana use, alcohol consumption, illicit drugs, viral infections and inherited conditions such as Down’s syndrome or Bloom’s syndrome among others [5].
Despite current advances in therapy of CL, it is still believed to have a rapidly progressive downhill course; only 20–23 % of children survive at 24 months [5, 6, 9, 10, 13–15].
In conclusion, we herein present a unique case of congenital leukaemia with initial aleukemic manifestation. So far, no validated diagnostic algorithm for “blueberry muffin syndrome” has been established [1]. We strongly suggest that after excluding intrauterine infections and hematologic dyscrasias, the next mandatory diagnostic step is to search for the underlying neoplastic disorder. Skin biopsy, immunohistochemistry staining along with cytologic and cytogenetic data are crucial to establish the origin of the infiltrate. DBC with percent blast cells, bone marrow aspirate with cytogenetic analysis and imaging studies such as chest X-ray, ultrasonography of abdomen and pelvis, brain tomography are necessary to assess any malignant systemic involvement heralded by typical cutaneous nodules [1, 10, 16, 17].