







Introduction
Morphea, also known as localized scleroderma, is a chronic, autoimmune disease of connective tissue, usually characterized by a typical clinical picture depending on the form of the disease, in the course of which, unlike in systemic scleroderma, there is no Raynaud’s phenomenon, sclerodactyly or ulcerations on the fingertips [1]. Although morphea and systemic sclerosis have been perceived as separate disease entities for years, they are still confused both by patients (which is a source of unnecessary stress) and doctors. This may be due to, in part, misunderstood terminology. However, the controversy around the morphea is not just about its naming. They also result from the division of this disease entity and the inclusion of rarer subtypes of morphea as separate diseases, including eosinophilic fasciitis. Discussions on morphea also involve diagnostic aspects, and over the years, have consistently been related to the possible treatment options. The paper attempts to present the debatable aspects regarding nomenclature, classification, diagnosis and treatment of morphea.
Controversy around the name
The term scleroderma was probably firstly used by Giovambattista Fantonetti in 1836 to describe a 30-year-old patient with extensive swelling of the skin of the limbs and trunk, accompanied by pustules. In a short period of time, hardening and browning of the skin was observed in this patient, while the areas of the nipples and face were spared [2]. The name scleroderma comes from Greek words (scleros, derma) and means “hard skin” [3]. Therefore, it is a term that describes a dermatological condition characterized by excessive fibrosis of the skin and adjacent structures [4]. However, this concept is a cause of confusion because it is used interchangeably with the term scleroderma, which is mostly perceived as a systemic sclerosis. Localized scleroderma is synonymous with morphea, i.e. a disease in which only the skin and adjacent structures are involved in locations different than those observed in systemic sclerosis. Ultimately, it seems that the use of the name scleroderma causes anxiety and needless stress among patients as well as among doctors of other specialties (hearing the term scleroderma they often assume that it is systemic sclerosis), which often causes unnecessary examinations and consultations. Therefore, in order to avoid misunderstandings, some experts in the field of localized scleroderma recommend the use of the term morphea [5, 6]. The name is also derived from Greek (form, structure) and indicates areas of induration located on the skin [5, 6].
Controversy over the division
Controversy in the case of morphea also concerns its division. The first classification was given by Tuffanelli and Winkelmann and they divided morphea into 3 subtypes: morphea, linear limited scleroderma and generalized morphea [7]. One of the first commonly used classifications is the division proposed by Peterson et al. in 1995, which distinguished five main types of scleroderma: plaque (including lichen sclerosus and atrophoderma of Pasini and Pierini), generalized, linear, bullous and deep (including eosinophilic fasciitis) [8]. This classification was debatable for two reasons. Firstly, it included lichen sclerosus, which according to the current classifications does not fall into the morphea spectrum, and secondly, it did not include the mixed form of morphea, which is currently estimated to occur in as many as 15% of patients [9]. In turn, Laxer and Zulian in 2006 presented the division of morphea, in which the mixed form appears, but the authors exclude the atrophoderma of Pasini and Pierini from the morphea spectrum [10]. In 2009, the classifications were modified by Kreuter et al., who proposed a division into five main clinical forms: localized, generalized, linear, deep and mixed [11]. This division is upheld by the European Dermatological Forum with the guidelines presented in 2017 [12]. Nevertheless, it still raises some controversy, e.g. with regard to the classification of progressive facial hemiatrophy or eosinophilic fasciitis (EF). The latter is perceived by some experts as a separate entity (discussed below).
The above-mentioned authors also define the main groups and subgroups differently. Most distinguish localized, linear and generalized forms as the main forms, but there is no consensus regarding, e.g. the disabling pansclerotic morphea. According to most recommendations, for example, by the authors of Polish, German or European guidelines, it is a generalized form [12–14], according to Laxer and Zulian and Fett et al., it belongs to the main forms of morphea [9, 10]. The same goes for the deep variety. According to Kreuter et al., Knobler et al. or Krasowska et al., it is one of the main forms of morphea, while according to Laxer and Zulian and Fett et al., it is a subtype of the restricted form. It should be noted that the use of the Laxer and Zulian classification is recommended by Japanese experts [15]. It is also worth emphasizing that in the literature, en coup de sabre and progressive facial hemiatrophy are sometimes collectively referred to as a craniofacial form or the head variant of morphea [9, 10, 16].
As already mentioned, some experts currently perceive EF as a separate disease entity [14, 17]. In 2014, specific diagnostic criteria for this disease were even proposed, but they have not been validated so far [17]. Significant distinguishing features of EF are: peripheral and tissue eosinophilia, peau d’orange, sulcus symptom, risk of restrictive lung disease [17]. According to the literature, even 63–93% of EF patients will present peripheral eosinophilia, and 60% – tissue eosinophilia [17–19]. It should be noted, however, that Zulian et al. demonstrated peripheral eosinophilia in 18% of children with plaque morphea [16], and Walker et al. presence of tissue eosinophilia and 18% of patients with MEP, 23% with a linear form and 15% with generalized morphea [20]. On the other hand, peau d’orange which is a sign of fascia involvement, can also be observed in deep morphea, pansclerotic morphea and graft versus host disease (GVHD) [21]. In the same diseases, the “groove” sign can also be observed, which is also found in up to 50% of patients with EF [21, 22]. Involvement of internal organs in EF is extremely rare, but there is a risk of restrictive lung disease. Only that this risk also applies to patients with pansclerotic morphea [21]. It has also been suggested that in morphea the skin involvement is focal, while in EF it is continuous. Nevertheless, continuity of skin involvement is also seen in pansclerotic morphea. Additionally, even in 65% of EF patients, coexistence with morphea plaques is observed [19]. Ultimately, in the opinion of the authors and in agreement with the opinion expressed, inter alia, by Fett et al., EF may be a severe form of morphea. Additionally, it should be borne in mind that the therapeutic regimens of EF do not differ significantly from those proposed in other forms of morphea, and therefore perhaps distinguishing this disease entity is unfounded.
In conclusion, it should be noted that the main features influencing the severity of morphea are: (1) the extent of the skin lesions, (2) the depth of the disease process, and (3) the craniofacial location.
Controversy around testing
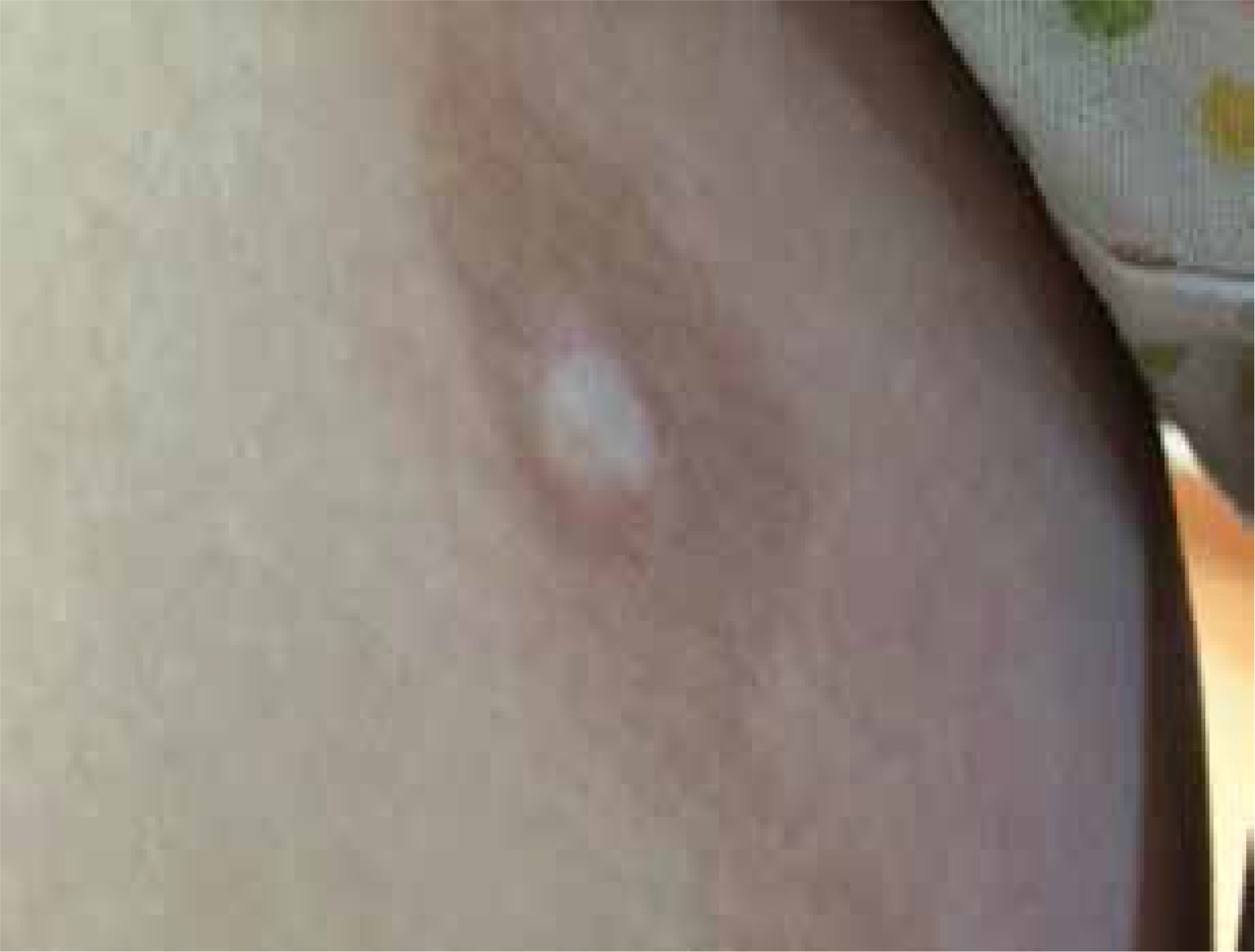
According to the current recommendations, as long as the patient presents with a classic clinical picture, there is no need to perform additional tests to confirm the diagnosis [12–14]. Taking a biopsy for histological examination should be reserved for cases of doubtful or unclear clinical picture [14]. As an invasive test, it can lead to a cosmetically unacceptable scar (Figure 1).
Figure 1
Hyperpigmentation in the area of previous morphea plaque with a visible scar in the centre after biopsy taken
No blood tests, including anti-nuclear antibodies (ANA), are highly specific and useful for the diagnosis of morphea [12–14]. Certain autoantibodies (e.g. ANA, anti-single-stranded DNA, anti-histone, anti-topoisomerase II, anti-RNP, anti-matrix metalloproteinase antibodies and rheumatoid factor antibodies) have been reported in patients with morphea [23, 24], but routine screening for these antibodies is only recommended to exclude the coexistence of other autoimmune connective tissue diseases (e.g. systemic scleroderma) [12].
The risk of laboratory abnormalities is greater in linear morphea and EF. The active phase of the disease may or may not be characterized by peripheral eosinophilia, accelerated erythrocyte sedimentation rate (ESR), increased levels of C-reactive protein (CRP) and muscle enzymes, including creatine phosphokinase (CPK) and aldolase [13, 14, 16, 25]. In addition, in the case of EF, the potential risk of coexistence of haematological disorders should be taken into account, which is estimated at 10–15% of all patients [19, 26]. Therefore, it is recommended to perform morphology and electrophoresis with immunofixation [12–14].
Magnetic resonance imaging (MRI) is an imaging study that has gained importance in recent years. It is recommended for all patients with craniofacial morphea in order to assess the central nervous system (CNS), regardless of the diagnosed neurological symptoms [12–14]. It is worth noting, however, that the usefulness of MRI in assessing the extent and depth (subcutaneous tissue, fascia, muscles) of the disease process, also in other areas of the body, is increasingly indicated. It can be used both as an aid in selecting the appropriate tissue biopsy site for histological examination and for monitoring the course of the disease and its response to treatment [27, 28]. Less frequently recommended imaging tests include computed tomography, high-frequency ultrasonography (HFUS), thermography, laser Doppler flowmeter, durometry and cutometer [12–15].
The importance of Borrelia burgdorferi (B. burgdorferi) in morphea is still controversial in the minds of many physicians. Such a relationship was first suggested by Aberer et al. in 1987 due to the similarity of skin lesions in the late stage of Lyme disease to morphea [29]. Over the years, many authors have tried to confirm this relationship with the use of various diagnostic methods, including silver staining, culture, ELISA, PCR, immunohistochemistry and focus-floating microscopy [9, 30–33]. Most of the results, however, did not confirm this. Ultimately, therefore, routine determination of antibodies to B. burgdorferi is not recommended, unless there is a reasonable clinical suspicion [12–14].
Controversy around the treatment
As it is well known, the essence of the treatment of morphea is to stop the active process and reduce its permanent damage, including especially the irreversible consequences of extra-cutaneous localizations, mainly regarding the musculoskeletal and nervous systems. Therefore, the time of treatment completion in patients with morphea, especially in deep and linear subtypes, is an extremely important issue. According to Florenz-Pollack et al., the presence of subcutaneous and cutaneous atrophy and hyperpigmentation lasting at least 6 months indicates the inactive disease [34]. However, it can be difficult to quantify this, especially in the case of linear and deep variants of the disease, like in morphea en coup de sabre. It seems controversial whether to consider it as a failure of therapy or as the natural course of the disease. What is more, the presence of such consequences should be always discussed with the patients. The typical course of morphea plaque from inflammation to atrophy takes approximately 3–5 years, while softening of sclerosis can be observed after an average of 2.7 years, and in the case of deep variants, even after 5 years [8, 18]. The studies show that the recurrence risk of morphea is high and may be associated with one-third to two thirds of patients also after completion of a full course of treatment with systemic medications [35, 36]. Moreover, it was noted that the recurrence rate after UVA1 phototherapy may be higher than after methotrexate [37]. Due to the recurrent course of morphea there is a need for long-term evaluations of patients especially suffering from linear and generalized types [36, 37].
Although we have had well-documented consensus in the field of treatment of morphea for several years, the issues of morphea therapy still arouse many discussions [12–14].
One of the controversial drugs is imiquimod, which modulates the immune response by inducing interferon γ (IFN-γ) and inhibiting transforming growth factor β (TGF-β) and thus has antifibrotic properties. So far, there are only a few case reports of the use of imiquimod in the topical treatment of morphea [38]. The efficacy of subcutaneously administered IFN-γ in morphea was investigated in a randomized, double-blind, placebo-controlled trial. Compared to placebo, there was no significant therapeutic effect [39]. Based on current data, this drug is not recommended for the treatment of scleroderma [12–14].
The therapeutic efficacy of D-penicillamine has been shown in a small number of cases, and there are still no controlled studies. Due to the questionable effectiveness and numerous side effects (including nephritic syndrome, headache, nausea, abdominal pain, diarrhoea) of D-penicillamine, it is also not recommended for the treatment of morphea [12–14].
Penicillin has been used for decades in the treatment of morphea and its potential efficacy has been explained by the link between B. burgdorferi infection and the development of morphological lesions of morphea. Penicillin was administered intravenously for 14–21 days, or alternatively, intravenous ceftriaxone was also used [11, 13]. In the literature, however, there are only reports of single cases of morphea in the treatment of which such therapy was used [40]. Möhrenschlager et al. presented the case of a 10-year-old female patient with linear morphea of the limbs who was treated with penicillin G intravenously at a dose of 5 MU three times a day for 10 days. Three months after a reduction in morphea, cohesion was observed, which correlated with a reduction in the thickness of skin fibrosis assessed by high-frequency ultrasound [40]. Although penicillin has an anti-inflammatory effect, no antifibrotic effects have been demonstrated so far [12, 13]. Due to the fact that the effectiveness of penicillin in morphea has not been finally confirmed, this form of therapy is not recommended at present [12–14].
Vitamin E is the name of a group of organic chemicals that include tocopherols and tocotrienols. α-Tocopherol shows the greatest biological activity. Vitamin E has antioxidant, anti-inflammatory and moisturizing properties. In 1996 Stein et al. conducted a study in which patients with systemic sclerosis in the generalized and limited form, compared to the control group, showed an increased level of oxidative stress [41]. Therefore, it has been suggested that antioxidants such as vitamin E may have beneficial effects in the treatment of this disease [42]. There are no data in the medical literature to support the efficacy of vitamin E, either oral or topical, in the treatment of patients with morphea. European recommendations, including German and Polish Dermatological Society recommendations from 2019, do not include vitamin E in the treatment of morphea [12–14].
The unsaponifiable fractions of avocado oil and soybean oil (ASU) are a complex mixture of avocado oil containing polyols, sterols, long-chain saturated hydrocarbons, squalene and tocopherol, and soybean oil, the main components of which are sterols and tocopherols [43]. This mixture was first produced in France and marketed as Piascledine. ASU increases the content of collagen and fat in tissues and causes a significant increase in the proportion of extractable components in relation to insoluble components with a significant increase in the activity of proteases and collagenoses in the serum. It is believed that the combination of soybean oil and avocado ingredients has a stronger effect than either alone [44]. Jabłońska used ASU in the treatment of patients with morphea, the mechanism of action has not been fully understood, it has been suggested that ASU has an inhibitory effect on some pro-inflammatory cytokines (including IL-1β, IL-6, TNF-α) and antioxidant [45]. There is still a lack of convincing data in the medical literature to support the effectiveness of ASU, and therefore, according to current guidelines, they are not recommended for the treatment of morphea [12–14].
Summary
In order to avoid unnecessary confusion on the part of both patients and doctors of other specialties, the authors postulate that localized scleroderma should be called morphea. The division of morphea still requires extensive consultations, while the authors of the article believe that EF is a particularly severe form of morphea. In localized morphea, there is no need for additional tests to confirm the diagnosis. Topical glucocorticosteroids, local calcineurin inhibitors and phototherapy are the mainstays of the treatment of active morphea in superficial lesions. However, treatment of severe types of morphea, such as linear (including ECDS), generalized and deep forms, requires the administration of methotrexate with or without systemic corticosteroids or mycophenolate mofetil.