







Introduction
Ataxia-telangiectasia (A-T) is an autosomal recessive genomic instability syndrome, resulting from a mutation of the Ataxia-Telangiectasia Mutated (ATM) gene. The gene, localized to 11q22.3-23.1, encodes for a high-molecular weight, predominantly a nuclear protein kinase which is a member of the large phosphoinositidil 3-kinase related protein kinase (PIKK) family [1]. The ATM kinase also plays many important cytoplasmic roles, phosphorylating numerous protein substrates and in mitochondrial respiration and energy metabolism. The enzyme is involved in maintaining the cell-cycle homeostasis and coordinates the cellular signalling pathways in response to DNA double-strand breaks (DSBs), genotoxic and oxidative stress [2]. The impact of the genetic background on the cellular nature of ataxia-telangiectasia is pleiotropic and the phenotype of the disease is complex and heterogeneous. A-T is a multisystemic disease characterized by neurodegeneration with progressively debilitating cerebellar ataxia, postural instability, choreoathetosis, dysarthria, problems with cough and swallowing and oculomotor apraxia. The extended A-T phenotype also includes interstitial lung disease, dermatological manifestations such as oculocutaneous telangiectasia and hormonal dysfunction, with growth retardation, insulin-resistant diabetogenic response and premature aging as well as infertility due to gonadal dysgenesis resulting from defective meiotic recombination. Affected patients also show a combined, humoral and cellular immunodeficiency with immune dysregulation, a predisposition to lymphoid malignancies and sensitivity to ionizing radiation which are markers of chromosomal instability [3].
The impaired activity of ATM kinase and defective DNA double-strand breaks reparation in A-T underlies the disturbed B and T cell homeostasis and a highly-variable immune deficiency [4]. The lymphocyte gene rearrangements, a tightly-controlled process resulting in clonal diversity and immunoglobulin class switch recombination (CSR) warranting the immune repertoire diversity as well as T cell proliferation and survival following T cell receptor stimulation are disturbed in A-T [1]. The affected patients show defective humoral immune response with hypogammaglobulinemia, predominantly low IgA and, to a lesser extent, also IgG levels, IgG subclass deficiency [5], low levels of antigen-specific antibodies to pneumococcal polysaccharide vaccine [6, 7], abnormal B cell neogenesis expressed as low KREC (κ-deleting recombination excision circle) copies [8], low B cell numbers, and impaired development of memory B cells due to antigen-dependent cognate B-T cell interaction [4]. Most A-T patients also show persistent T cell lymphopenia, with low numbers of CD4+ and CD8+ naive T cells and both the CD4+ and CD8+ CD45RA+ : CD45RO+ ratios less than one [5] as well as low TREC (TCR rearrangement excision circle) copies and abnormal TCR-Vβ repertoire in spectratyping with different degree of clonality [7, 9], reflecting thymus dysfunction in generating naive T cells.
A rare but well-recognized manifestation of immune deficiency in A-T is the formation of non-infectious granulomas. The developmental abnormalities of B and T cell compartments and impaired immunosurveillance resulting in defective immune response to microbial antigens as well as inappropriate immune regulation and innate immune response-driven inflammation are the leading causes of granulomatosis in A-T.
Aim
The aim of the study was to investigate the clinical, pathological and immunological features of cutaneous and systemic granulomatosis in A-T.
Material and methods
We retrospectively reviewed medical records of 8 A-T children, aged from 2 to 18 years (median age: 10 years) who had been diagnosed and treated in our university clinic for paediatric pneumonology, allergology and clinical immunology. We analysed the clinical manifestations of cutaneous and visceral granulomas in affected patients. Herein we present laboratory investigations in 1 A-T patient with cutaneous and systemic granulomatosis, including serum biochemistry, immunoglobulin assessment (an immunoturbidimetric method, Beckman Coulter, USA), and flow cytometric peripheral blood lymphocyte immunophenotyping. For the purposes of evaluating deep organ localization of granulomas, the magnetic resonance imaging (MRI) of the chest and the abdominal cavity was performed in 2 patients. In 2 patients, a biopsy and the histopathological examination of the cutaneous granulomas and in 1 patient examination of the laryngeal granuloma during laryngoscopy and revision of the tracheostomy were carried out. Informed consent was obtained for all parents / legal guardians of the children studied.
Flow cytometric peripheral blood lymphocyte immunophenotyping was performed according to the standard methods described by Piatosa et al. [10] and Schatorje et al. [11].
Peripheral venous blood cells were labelled with the following fluorochrome-stained monoclonal antibodies: anti-CD45 FITC (fluoresceineisothiocyanate), anti-CD14 PE (phycoerythrin), anti-CD19 PE, anti-CD19 PerCP (peridinin chlorophyll protein), anti-IgM FITC, anti-IgD FITC, anti-CD38 APC (allophycocyanin), anti-CD27 PE, anti-CD21 FITC, anti-CD3 FITC, anti-CD4 FITC, anti-CD45RA FITC, anti-CD127 FITC, anti-CD185 FITC, anti-CD8 PE, anti-CD16+CD56 PE, anti-CD25 PE, anti-CD27 PE, anti-CD31 PE, anti-CD45RO PE, anti-CD3 PerCP, CD197 PerCP, anti-CD4 APC and anti-CD8 APC (all Becton-Dickinson Biosciences, USA).
The acquisition of cells and analysis were carried out with the use of flow cytometer FACSCanto and FACSDiva software (Becton-Dickinson, USA).
With sequential gating on biparametric scattering CD45hiCD14- lymphocytes, the following lymphocyte populations were identified: among B cells: memory B cells CD19+CD27+, naïve B cells CD19+CD27-, non-switched memory B cells/marginal zone B cells CD19+CD27+IgD+, switched memory B cells CD19+CD27+IgD-, transitional B cells CD19+CD38hiIgMhi, plasmablasts CD19+CD38+IgM-, and immature B cells CD19+CD21lo, CD19+CD21loCD38lo, and among CD3+T cells: T helper cells CD3+CD4+, cytotoxic T cells CD3+CD8+, natural killer (NK) cells CD3-CD16+/CD56+, NK-T cells CD3+CD16+/CD56+, naive T helper cells CD3+CD4+CD45RA+CD27+, terminally differentiated T helper cells CD3+CD4+CD45RA+CD27+, central memory T helper cells CD3+CD4+CD45RA-CD27+, effector memory T helper cells CD3+CD4+CD45RA-CD27-, naive cytotoxic T cells CD3+CD8+CD45RA+CD197+CD27+, terminally differentiated cytotoxic T cells CD3+CD8+CD45RA+CD197-CD27-, central memory cytotoxic T cells CD3+CD8+CD45RA-CD197+CD27+, effector memory cytotoxic T cells CD3+CD8+CD45RA-CD197-CD27-, recent thymic emigrants CD3+CD4+CD45RA+CD31+, regulatory T cells CD3+CD4+CD25++CD127-, and circulating memory T cells CD3+CD4+CD45RO+CD185+.
Results
In the 2 A-T patients studied, one 2-year-old and one 12-year old, mild maculopapular skin lesions of varying intensity localized on the cheeks and forearms were observable. In the other 2 patients, a 13-year-old boy and a 10-year-old girl, cutaneous granulomas clinically presented as disseminated skin nodules and ulcerated erythematous plaques localized on the face and on trauma-prone areas of upper and lower extremities in the regions of joints and on palms and feet. The lesions were scaly and ulcerated, covered with crusts and scarring (Figures 1 A–C).
Figure 1
A – An MRI of the abdominal cavity and the retroperitoneal space with contrast medium, transverse section, showing a markedly enlarged spleen (its dimensions 17.3 × 8.5 cm, length x width, respectively) with non-homogenous nodular remodelling of its parenchyma. These numerous infiltrates of different size (from 5 to 60 mm) exhibit hypointense signals in T1 and T2- dependent sequences. B – An MRI of the chest with contrast medium, transverse section, showing mediastinal and perihilar lymphadenopathy and numerous small (size from 4 to 6 mm of a diameter) perilymphatic nodules, localized subpleurally and surrounding bronchovascular bundles. In the right middle lobe (segment 5) pericardial consolidations in the lung parenchyma with fibrotic strands, accompanied by irregular thickening of the peribronchium of the bronchial tree and peripheral bronchiectases are visible. C – An MRI of the neck, without contrast medium, frontal section, showing a thickening of the glottis, the aryepiglottic folds, the vestibular folds, and the vocal cords. At the level of the laryngeal inlet and upper part of the vestibule, the patency of the larynx is significantly reduced (to 2 mm of a diameter) with a slit-like upper part of the piriform sinus. In the oropharynx, hypertrophy of the lymphoid tissue is visible
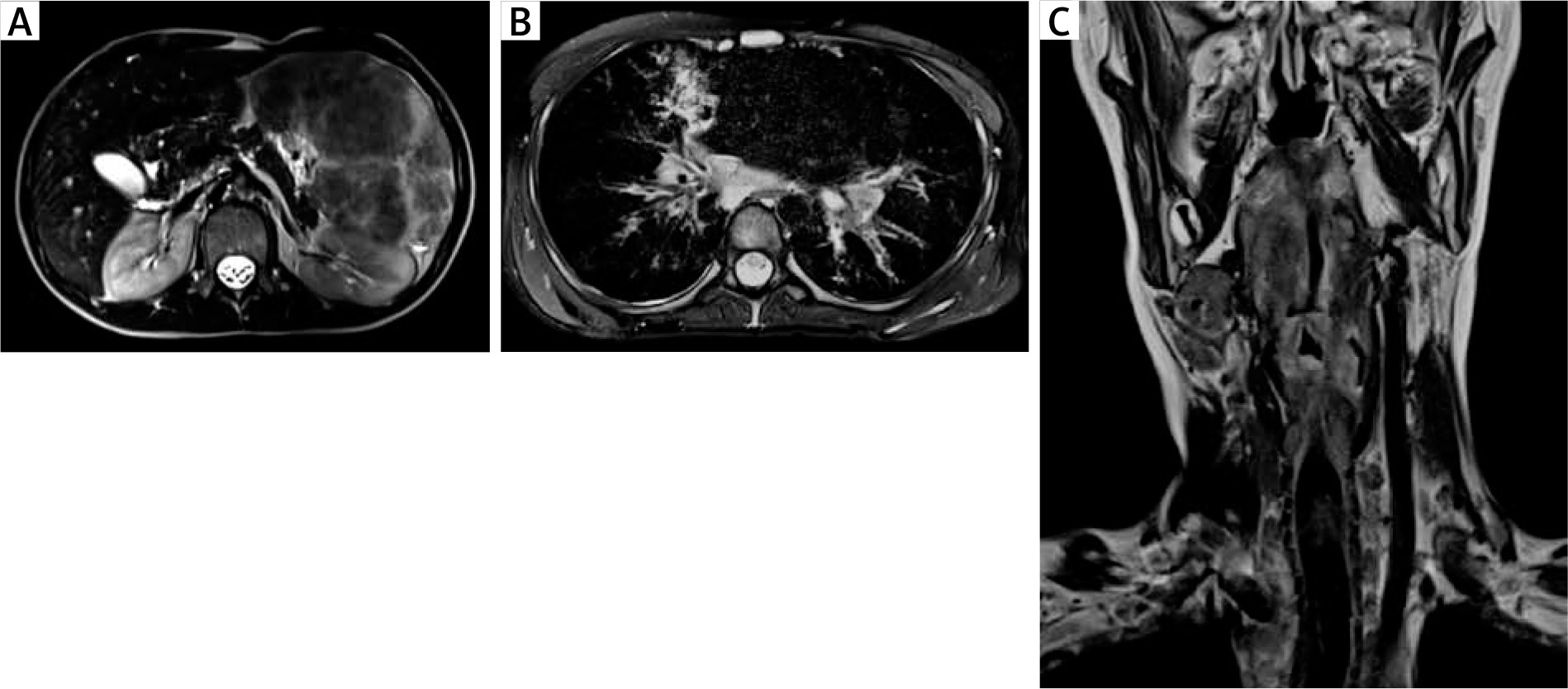
In 1 A-T patient, visceral granulomatosis involved the lungs, the spleen, the liver and the larynx. In the larynx, its almost total obstruction by granulomatous infiltrations in the area of laryngeal inlet and vestibule, assessed in MRI and in laryngoscopy as well as clinical symptoms of increasing dyspnoea and stridor led to the need for tracheostomy in emergency conditions (Figures 2 A–C). In this patient, persistent leukopenia, anaemia and mild thrombocytopenia, accompanied by a decreased relative value and absolute count of lymphocytes and a borderline neutrophil count in the differential white blood count (WBC) were observable. A characteristic hyper-IgM phenotype with a low serum IgG level and a total IgA deficiency and a markedly increased level of IgM correlated with the clinical severity of cutaneous and visceral granulomatosis (Table 1).
Figure 2
A – Tuberoid deformations of the fingers. B – Extensive, exophytic, scarred granulomatous lesions in the area of the elbow joint. C – Ulcerated, crusty and bleeding lesions of the knee
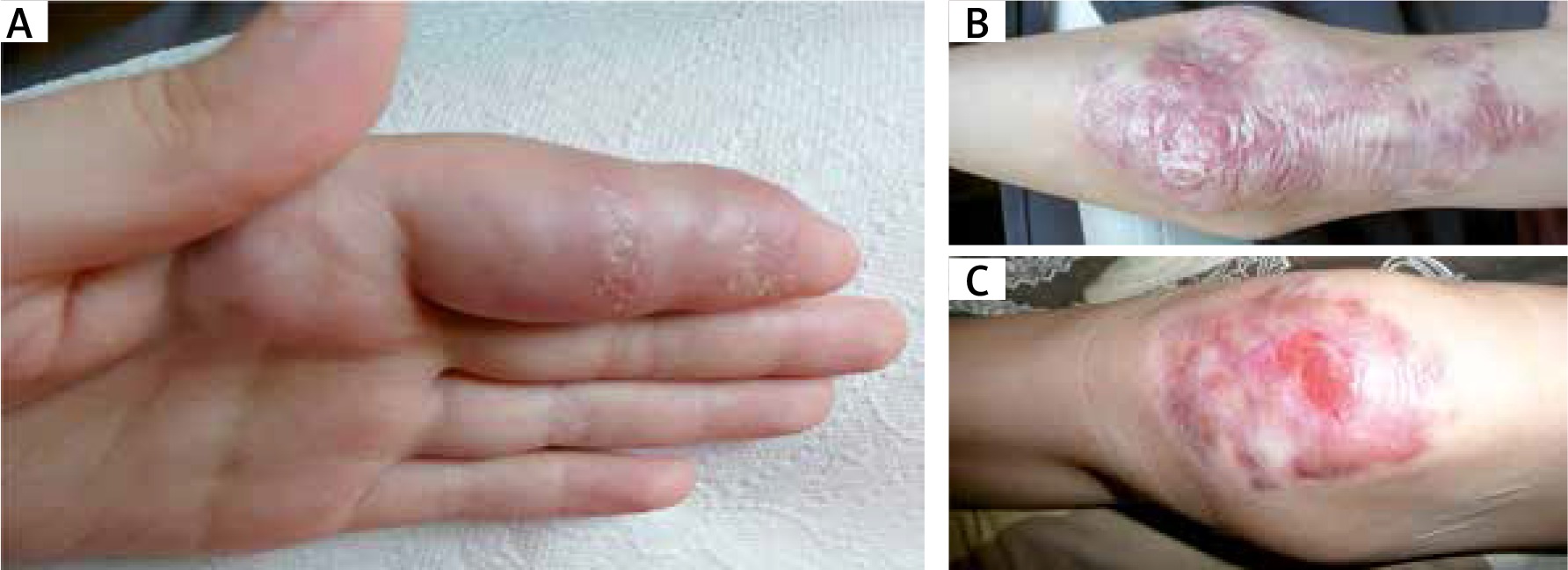
Table 1
Immunological data of an A-T patient with cutaneous and systemic granulomatosis
In the same patient, peripheral blood lymphocyte flow cytometric immunophenotyping showed a markedly reduced CD19+ B cell subset, with an almost total lack of CD19+CD27+sIgD- switched memory B cells and a predominance of IgM-only memory B cells. Among the T cells, a markedly reduced number of CD3+CD4+CD45RA+ naive T helper cells and CD3+CD8+CD45RA+ cytotoxic T cells with a memory phenotype and with a predominance of CD3+CD4+CD45RA- T cells was observable. The T cell receptor repertoire was skewed towards the γδ T cells and it was accompanied by an increased number of NK cells (Table 1).
In this patient, histological examinations of the biopsies of the skin and the larynx were performed and they showed features of non-caseating, non-infectious granulomas with CD4+ : CD8+ T lymphocyte ratio << 1 and negative results of immunochemistry-based assays towards biomarkers of oncogenesis (Table 2). Ziehl-Neelsen staining of the skin and epiglottis biopsies to identify acid-fast Mycobacteria and polymerase chain reaction (PCR) assay for Mycobacterium tuberculosis were negative. Examinations of EBV (Epstein-Barr virus), CMV (cytomegalovirus), HBV (hepatitis B virus), HCV (hepatitis C virus), Mycoplasma pneumoniae, HSV (herpes simplex virus) 1 and 2, HHV (human herpes virus) 6 and 7, enterovirus, adenovirus, parechovirus, hPV (human parvovirus) B19 as well as HIV (human immunodeficiency virus) in blood proved negative.
Table 2
Histological features of cutaneous and laryngeal granulomas in A-T
In all the A-T patients studied, intravenous immunoglobulin replacement therapy was administered, but no improvement of granulomatous lesions in 4 affected children was observed. In the most severely affected boy, a course of topical (clobetasol) and systemic corticosteroids (methylprednisolone) was also conducted; however, the clinical effect on both cutaneous and visceral granulomas was poor.
Discussion
Granulomatous skin lesions are a rare, but well-recognized manifestation of primary immunodeficiency diseases (PIDs) and affect first of all patients with disturbed B and T cell compartments in A-T [12, 13], with the vast majority occurring in three diseases, namely A-T, common variable immunodeficiency (CVID) and severe combined immunodeficiency (SCID) with the first two diseases each being responsible for more than 30% of the cases [14]. However, granulomatous skin disease was also observed in a variety of PIDs, namely in syndromes associated with combined immunodeficiencies, such as Nijmegen breakage syndrome (NBS) [15–17] and Wiskott-Aldrich syndrome [18], severe combined immunodeficiencies due to RAG (Recombination Activation gene) deficiency [19], JAK3 (Janus kinase 3) deficiency [20], and Artemis deficiency [21], HLA class I deficiency syndrome [22], deficiencies of the innate immunity, namely chronic granulomatous disease [23] and chronic autoinflammatory disorders, such as Blau syndrome [24].
Cutaneous granulomas are typically accompanied by visceral granulomatosis in CVID, most frequently localized in the lung as granulomatous lymphocytic interstitial lung disease (GLILD) [25], in the spleen, in the liver [26], and in the intestine [27], but we had also previously reported pulmonary lymphomatoid granulomatosis in a paediatric patient with Griscelli syndrome type 2 [28]. It is worth noting, that in A-T, in contrast to CVID and, to a lesser extent to SCID, cutaneous granulomatosis is uncommonly accompanied by visceral granulomas and this is the first report of severe, multiorgan involvement of the granulomatous disease in an A-T patient.
The immunopathogenesis of these non-infectious, non-caseating granulomas is not fully understood, but in the setting of combined B and T cell immune deficiency being a common immunological denominator, inappropriate immune regulation has been proposed as playing a major role in the granulomatous pattern [13, 14]. All 4 of our A-T patients showing cutaneous granulomatosis, in particular the one with its systemic manifestation, demonstrated markedly skewed development of B and T cell populations. Lymphopenia, with an almost total lack of CD19+ B cells, in particular of a naive CD19+CD27-sIgD+ B cell subset was observable. For the patient with visceral granulomatosis, a hyper-IgM immunophenotype with a decreased IgG level and IgA deficiency with a normal or elevated IgM level in serum was a particular characteristic. The immunological profile was accompanied by a profound deficiency of CD19+CD38hisIgM– plasma cells and CD19+CD27+sIgD- switched memory B cells with an increased number of CD19+CD27+sIgD–sIgM+ IgM only memory B cells, reflecting a defective Th2-driven immunoglobulin isotype switching in B cells in germinal centres. Among CD3+ T cells, a deficiency of naive both T helper and cytotoxic T cells was shown, with aberrant TCR (T cell receptor) repertoire and an increased number of T cells bearing a γδ receptor, which was accompanied by a high number of NK cells.
The pathogenesis of granuloma formation in this immunodeficiency setting is not fully understood, but it has been hypothesized that these profound lymphocyte developmental disturbances, absence of B cells and naive T cells with an inappropriate T cell receptor repertoire and unopposed activity of γδ T cells and NK cells result in immune dysregulation of macrophages [29, 30]. Macrophages play a critical role in the initiation and maintenance of inflammation and the granulomatous response. They are activated by various proinflammatory mediators including interferon γ (IFN-γ) secreted by NK cells and γδ T cells which are abundant in A-T patients with granulomatous skin disease [12, 13] and in our patient with visceral granulomatosis. Moreover, a deactivation of macrophages and a resolution of granulomas require anti-inflammatory cytokine interleukin (IL)-10, which is, in normal conditions, produced by Th2 cells. Thus, not only is the induction of granuloma formation enhanced by NK and skewed Th1 population of γδ T cells, but also a Th2-related counterregulatory mechanism of granuloma resolution are deficient in A-T. This immune dysregulation of macrophages, due to impaired lymphocyte neogenesis proposed as the predominating immunopathological phenomenon in cutaneous granulomatosis may also play a role in the pathogenesis of granulomatous disease in other organs.