







Palmar-plantar pustulosis (PPP) is a chronic inflammatory disease characterized by aseptic pustules of erythematous keratotic lesions primarily involving the palms and soles of the hands [1]. According to the existing diagnostic criteria for SAPHO syndrome, when a patient presents with palmoplantar pustulosis combined with osteoarticular involvement, then SAPHO syndrome is diagnosed. The common sites of bone and joint involvement in SAPHO syndrome are the anterior chest wall (ACW), the midshaft bones, and the long bones of the extremities [2]. There are no reports of PPP patients with combined hip involvement diagnosed as SAPHO syndrome. Here, we report the case of PPP combined with hip involvement, which was treated with JAK inhibitors combined with bisphosphonates with significant improvement in symptoms.
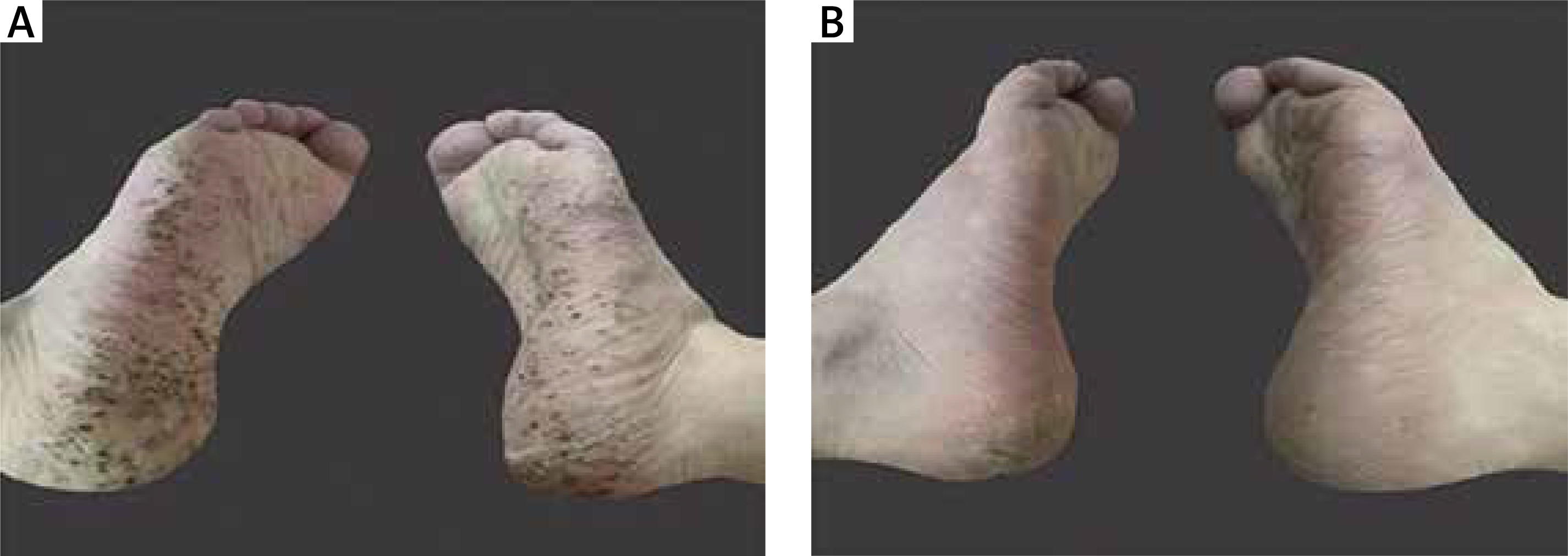
A 36-year-old man with a 7-year history of left inguinal pain was treated intermittently with oral nonsteroidal anti-inflammatory drugs (NSAIDs) for pain relief. Three years ago, plantar pustules appeared, and a diagnosis of palmoplantar pustulosis (PPP) was made. The patient denied the presence of anterior chest pain during the course of the disease, the use of hormones, and the use of alcohol, tobacco, and other bad habits. Recent hip computed tomography (CT) suggested: localized cortical discontinuity and slight collapse of the left femoral head, and multiple necrotic lesions with peripheral osteosclerosis. Hip magnetic resonance imaging (MRI) compression fat sequence suggested: bone marrow oedema of the left femoral head (Figure 1). Laboratory tests showed that HLA-B27 was negative; C-reactive protein 13 ↑ (0–10 mg/l); and blood sedimentation 25↑ (0–15 mm/h). The patient’s pain improved after 3 days of bisphosphonate (zoledronic acid 4 mg qd) intravenous treatment, followed by oral tofacitinib 5 mg bid treatment, and the patient’s symptoms of the rash improved significantly after 1 month of treatment (Figure 2).
Figure 1
A, B – Hip CT suggested: localized cortical discontinuity and slight collapse of the left femoral head, and multiple necrotic lesions with peripheral osteosclerosis. C, D – Hip MRI compression fat sequence suggested: bone marrow oedema of the left femoral head
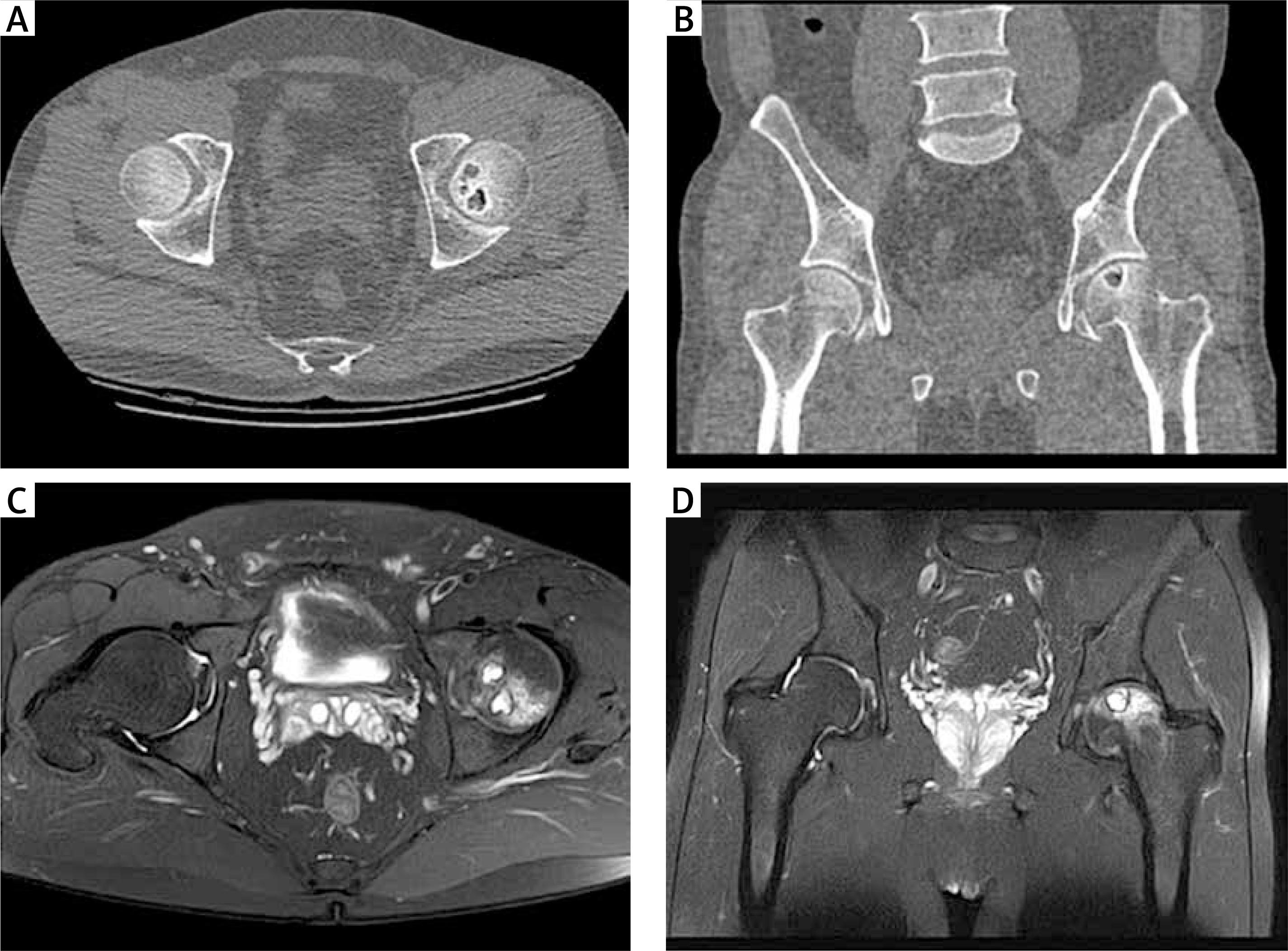
Figure 2
A – Prior to treatment, the patient had a scattered rash on the soles of the feet. B – After tofacitinib and bisphosphonate treatment, the patient’s plantar lesions improved significantly
Palmoplantar pustulosis is a rare, chronic, relapsing inflammatory disease that is the most common cutaneous manifestation of the SAPHO syndrome [3]. Osteitis and osteophytes are the core clinical manifestations of SAPHO syndrome, and when typical bone lesions are located at characteristic target sites, (e.g., sternoclavicular joints (93.0%), costovertebral joints (87.0%), sternoclavicular joints (84.5%), spine (33%), and sacroiliac joints (16.6%), among others [3]), the diagnosis of SAPHO syndrome is not difficult. However, if atypical sites are involved in patients with dermatologic disease, (e.g., mandible (2–10%) [4], skull (4.5%) [5] etc.), making the diagnosis is often challenging. The hip is a rare site of involvement in SAPHO syndrome, with a prevalence of only 3% [6].
SAPHO syndrome and spondyloarthritis all have the potential to involve hip joint lesions, and we need to identify such diseases in a timely manner. Approximately 92.9% of SAPHO and 97.7% of SpA patients with MRI pressure-lipid sequences are capable of developing bone marrow oedema and patchy bone oedema [7]. A study by Iida et al. found that bone marrow oedema was highly correlated with subsequent femoral head collapse and that bone marrow oedema was considered a marker of potential progression to advanced osteonecrosis [8]. Some reports suggest that hip involvement in AS may be associated with synovitis or attachment point inflammation [9]. Hip MRI manifestations in patients with AS may show joint effusion and synovial enhancement due to synovitis in addition to bone marrow oedema [10]. The MRI of the hip joint in our patient showed bone marrow oedema without HLA-B27 positivity and other AS manifestations, so we hypothesized that this patient presented with palmoplantar pustulosis combined with hip arthropathy, a special type of SAPHO syndrome.