







Progressive cribriform and zosteriform hyperpigmentation (PCZH) is a late-onset disorder of pigmentation with a zosteriform distribution [1]. It is considered to be the localized form and late onset of linear and whorled nevoid hypermelanosis (LWNH) [2]. Clinically, PCZH is characterized by cribriform macular hyperpigmentation, usually on the trunk [2].
A 24-year-old male presented with a Blaschkoid, cribriform, brown macule on the trunk for 16 years. It appeared at the age of eight and has been slowly progressing. He was otherwise in good health without other skin abnormalities. No prior injury was reported to the affected skin. His parents were nonconsanguineous and denied any history of neurofibromatosis or similar eruptions. On examination, there was discrete cribriform hyperpigmentation on the trunk with a Blaschkoid distribution (Figure 1). The lesions were pigmented homogeneously. His hair, nails, and mucosae showed no abnormality. Routine blood tests, urinalysis, liver and renal function tests, and electrolytes were all within normal limits. Skin biopsy specimens from a representative lesion showed elongated epidermal rete ridges and an accumulation of pigment at the tip of rete ridges (Figure 2). There were a few melanophages in the dermis. The plethora of clinicopathological conditions is consistent with the diagnosis of PCZH. No treatment was given, and the lesion remained unchanged until now.
Figure 1
The uniformly pigmented macules arranged in cribriform, S-shaped, or Blaschkoid configurations following the Blaschko’s lines on the back (A) and right flank (B)
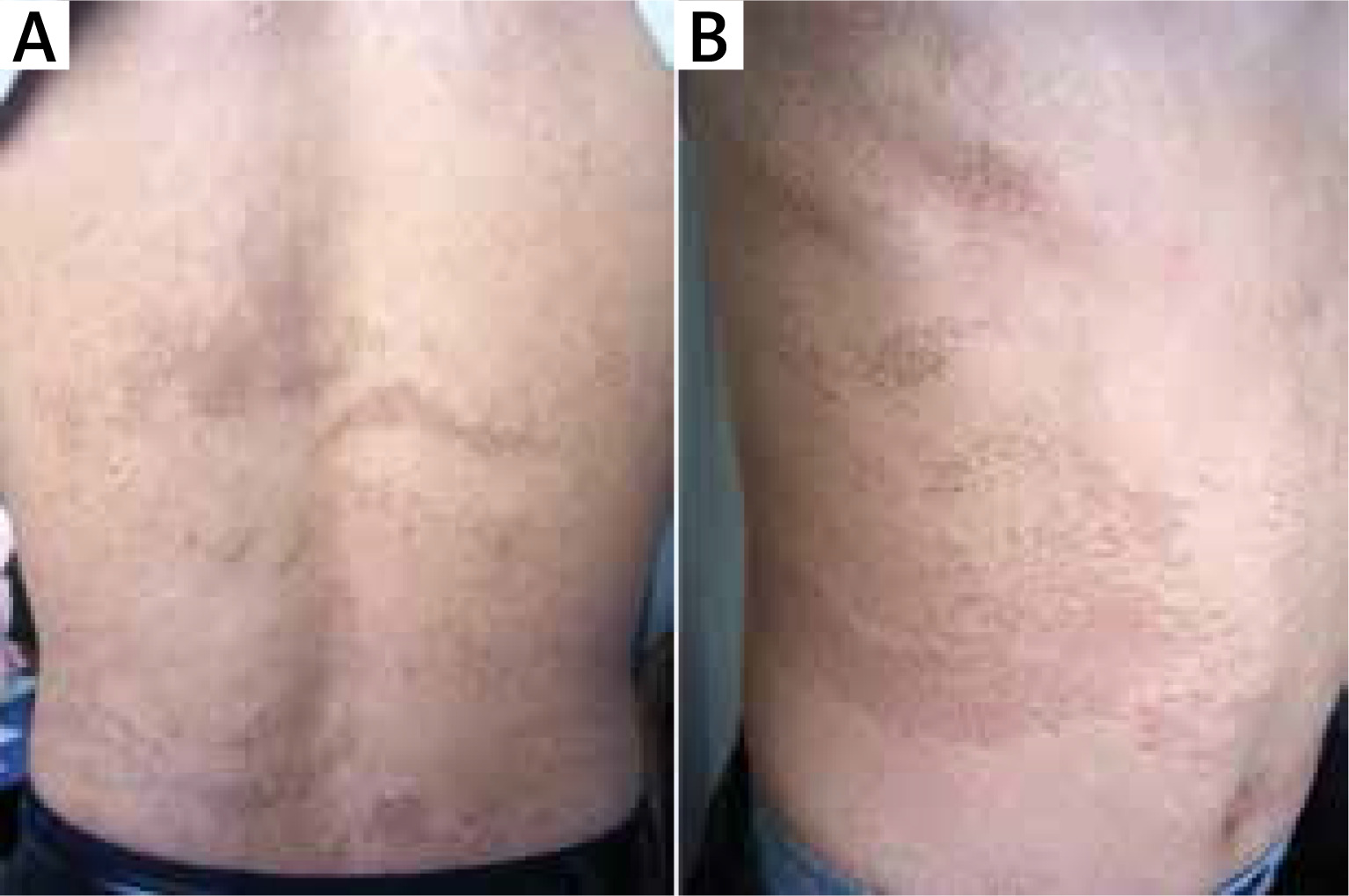
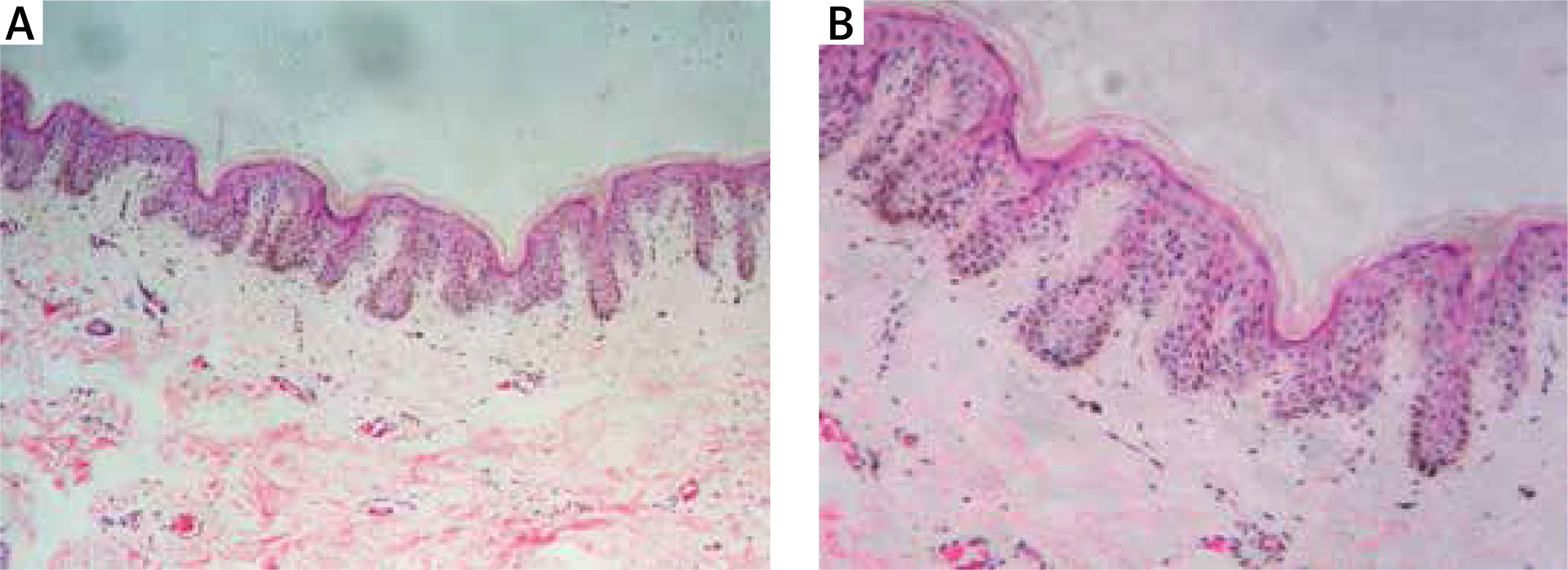
Figure 2
Elongated epidermal rete ridges (A, HE 100×) and an accumulation of pigment at the tip of rete ridges in the basal layer. There were a few melanophages in the dermis (B, HE 400×)
PCZH is asymptomatic cribriform hyperpigmentation distributed along Blaschko’s lines. It was first reported by Rower et al. without a defined incidence [1]. PCZH has also been referred to using other terms including “zosteriform lentiginous nevus”, “reticulate hyperpigmentation of Iijima”, “zebra-like hyperpigmentation in whorls and streaks’’ and ‘‘reticulate hyperpigmentation distributed in a zosteriform fashion”. There is no sex preference, and the mean age of onset is estimated to be 14 years [3].
PCZH lesions are characterized by uniform, cribriform macular hyperpigmentation in a zosteriform or Blaschkoid distribution. It often has a late onset with gradual extension, and the trunk is the predilection site of involvement. There is typically evidence of preceding trauma or inflammation. PCZH may occur with extracutaneous abnormalities, including mental retardation, polythelia, accessory mammary tissues, kidney, and urinary tract malformation [2–4]. Skin biopsy of the lesion in PCZH reveals increased pigmentation of the basal layer without epidermal melanocytosis. Melanophages may rarely be found in the dermis [3, 5]. Similar to Dowling-Degos disease, our patient unprecedentedly showed elongated epidermal rete ridges and hyperpigmented tips of rete ridges.
A set of diagnostic criteria was proposed for PCZH with five diagnostic thresholds [4]: (1) uniformly tan cribriform macular pigmentation along the Blaschko’s lines; (2) basal layer hyperpigmentation and absence of nevus cells; (3) excludes postinflammatory hyperpigmentation with the absence of preceding injury, inflammation or other eruptions; (4) starts in young adulthood (usually in the second decade of life) with gradual extension; and (5) devoid of other related skin or systemic abnormalities.
Differential diagnosis of PCZH includes pigmentary disorders occurring along the Blaschko’s lines, such as LWNH, café-au-lait spots, incontinentia pigmenti (IP), and Becker’s naevus. LWNH shows a similar clinical presentation but shows asymmetric hyperpigmentation in streaks and swirls rather than localized eruptions, as in PCZH. The concomitant congenital anomalies and the onset age at birth in LWNH did not presented in our patient [2, 3]. Macular hyperpigmentation was cribriform in our patient, without showing continuous tan-brown pigmentation in café-au-lait spots. In addition, the Blaschkoid distribution makes the diagnosis of café-au-lait spots unlikely [6]. The absence of a vesicular or verrucous stage and the female preponderance helps to exclude the diagnosis of IP [3]. Becker’s naevus presents with hyperpigmented macules. The lack of the cribriform configuration and hypertrichosis distinguish the abnormality noticed in our patient from Becker’s naevus [3]. Other differential diagnoses are acanthosis nigricans, confluent and reticulated papillomatosis, postinflammatory hyperpigmentation, erythema ab igne, and nevus spilus. They can all be ruled out by the history, classical clinical presentation, and typical pathologic changes in our patient.
The exact pathogenesis of PCZH has not yet been clarified. The distribution following the Blaschko’s lines in PCZH adds fuel to this prevailing theory that somatic mosaicism may give rise to two populations of melanoblasts with different potentials for melanogenesis [3]. However, PCZH with associated mosaicism has not been described. Currently, no effective treatment modalities exist for PCZH.
Our patient showed typical presentations of PCZH, except for pathological changes of elongated epidermal rete ridges and an accumulation of pigment at the tip of rete ridges. Therefore, PCZH is a histologic mimicker of Dowling-Degos disease and should be excluded pathologically before PCZH is diagnosed.