





The human body is colonized by millions of microorganisms, which are not only ‘passive residents’ but also play an extremely important role in maintaining homeostasis [1]. The term ‘microbiota’ refers to all the microorganisms living in a particular environment. The commonly used term ‘microbiome’, on the other hand, is defined as the ensemble of genomes of the microbiota considered in terms of the modulatory factors and interactions among them. The above term was coined microbiome by Joshua Lederberg in 2001. Another important term is ‘dysbiosis’, which means alterations in the number, composition and function of the microflora [2]. Variably intensified imbalance of commensal flora is observed virtually in every pathology and is always accompanied by dysfunction of the immune system [3–5]. Therefore, a detailed understanding of the relationships between the microbiome and functioning of individual systems allows implementing the most appropriate treatment, both pharmacological and dietary.
THE GUT MICROBIOME
The human gastrointestinal (GI) tract is colonized by more than 100 trillion bacteria, both commensal and pathogenic, affecting homeostasis. The gut microbiome consists of four main phyla, which include Bacteroides (23%), Firmicutes (64%), Actinobacteria (3%) and Proteobacteria (8%) [6, 7]. It is worth noting that the composition of microflora varies depending on the GI segment and age of an individual [6]. The lowest number of bacteria is found in the stomach due to its low pH, mostly different species of the genera Lactobacillus, Veillonella and Helicobacter [7]. The major bacterial families found in the small intestine include Bacilli, Actinobacteria, Streptococcaceae, Actinomycinaeae and Corynebacteriaceae while Bacteroidetes predominate in the large intestine [7, 8]. The composition and activity of the gut microbiome is affected by diet, lifestyle, environment, and genetic conditions [9]. The gut microbiome plays an important role in nutrient absorption, synthesis of vitamins (K, B1, B6, B12, folic acid), amino acids, enzymes, and production of short-chain fatty acids (SCFA). For instance, cellulose and pectin are converted into SCFA and simple sugars. By-products, including acetate, propionate and butyrate, are involved in the production of energy in cells, strengthen the integrity of the epithelial barrier and play an essential role in immunomodulation and protection against pathogens. Scientific studies have shown that up to 10% of the energy supplied can come from the above metabolic processes, e.g. butyrate is an important energy source for cells in the large intestine. Furthermore, butyrate stimulates the production of cathelicidin, which shows antibacterial properties [10]. The GI microflora plays an equally important role in maintaining homeostasis of the immune system. The immune system of the GI tract has an exceptional ability to create immune tolerance to the ever-changing and huge microbiome. Moreover, it is capable of producing an effective immune response to pathogens. Therefore, the largest number of immune cells is found in the places colonised by commensal organisms, such as the GI tract or skin. Furthermore, the microbiome strengthens the intestinal epithelial integrity, stimulates the proliferation of enterocytes and mucin, affecting ‘the barrier immunity’ [11, 12]. The barrier formed by the epithelium, mucus, immunoglobulin A (IgA), antimicrobial peptides and immune cells is organized around hyperglycosylated mucin 2 (MUC2), which provides static protection and constrains the immunogenicity of gut antigens via the dendritic cells. Moreover, the intercellular junctions and the mucus produced by the goblet cells form an important barrier limiting the translocation of microbes [13]. Scientific studies have shown that the gut microbiome affects the production of secretory IgA antibodies and cationic antimicrobial peptides (CAMPs) by the epithelial cells, which additionally maintain the function of the mucosal barrier [14, 15]. The production of regenerating islet-derived protein 3-gamma (Reg III-γ), the expression of which begins immediately after birth, is controlled by the flora in a myeloid differentiation primary response gene 88 (MyD88)-dependent manner and has the direct bactericidal effect on Gram-positive pathogens [16, 17]. Moreover, antimicrobial peptides have the potential to maintain the microbiome-intestine barrier [18]. Commensal bacterial antigen-specific IgA is produced by the dendritic cells in the GI tract [19]. IgA interacts with B and T lymphocytes in Peyer’s patches [19]. Of note, IgA responses lack classical memory characteristics and can address the changes in the microflora composition [20].
THE LUNG MICROBIOME
In recent years, the belief that the lungs are “sterile” has been denied, although the role of commensal organisms inhibiting the lungs has not been explicitly documented. It is known, however, that during micro-aspiration of nasopharyngeal content or in cases of gastro-oesophageal reflux to the alveoli, microorganisms are constantly “supplied” [21, 22]. Studies in animals and healthy volunteers have demonstrated that the composition of the lung microbiome is different from that of the oral and gut microbiome [23, 24]. The exact composition of the lung microbiome is difficult to be determined, as difficulties in ideal sampling of materials significantly limit its reliability. The key factor affecting the reliability of bronchoscopic samples is their contamination with the nasopharyngeal bacterial flora. The difficulties in collecting the microbial material, representing a reliable composition of the lower respiratory tract microbiome, are lesser in intubated patients [24]. According to some reports, only highly invasive methods, such as an open lung biopsy, allow the examiner to obtain a reliable testing material [25]. The lung microbiome is a dynamic ecosystem that consists of a variety of commensal microorganisms, mostly bacteria whose distribution depends on the part of the respiratory system. Moreover, there is a complex integrity between the upper and lower respiratory tract microbiomes, which may be confirmed by the fact that Firmicutes, Bacteriodetes, Proteobacteria, Fusobacteria and Actinobacteria predominate in healthy lungs [26–30]. It is worth stressing that the density of the pulmonary microbiome is low, ranging from 103–105 CFU g−1 tissue under physiological conditions [31, 32]. Animal studies have revealed that the airway flora formed during the first weeks of life is crucial for the development of a properly functioning immune system. This flora affects the Helios (−) regulatory T cells (Tregs) and reduces the susceptibility to allergic respiratory diseases [33]. Early formation of the microbiome also affects the stability of microflora in the upper respiratory tract and lower susceptibility to infectious diseases [34]. Numerous observations have shown a close relationship between the upper and lower respiratory tract microbiomes, especially in acute and chronic inflammations, such as obstructive pulmonary disease or cystic fibrosis [35–38]. The above correlation is associated with microaspiration, especially in cases of gastro-oesophageal reflux disease or disorders of airway cleansing [35]. The respiratory microflora induces the differentiation of peripheral Tregs, which are essential for controlling type 2 immune responses. Experimental studies have revealed a significant relationship between the natural killer T cells (NKT cells) and the microbiome [39, 40]. In the absence of airway microflora, increased numbers of eosinophils and type 2 T helper (TH2) lymphocytes have been observed in animal lungs [39, 40]. The lung microbiome is believed to modulate the expression of immune cell genes by affecting an increase in interleukin-5 (IL-5), interleukin-10 (IL-10), interferon γ (IFN-γ), C-C motif chemokine ligand 11 (CCL11) and toll-like receptor-4 (TLR4)-dependent responses of the lung macrophages [41] (Figure 1).
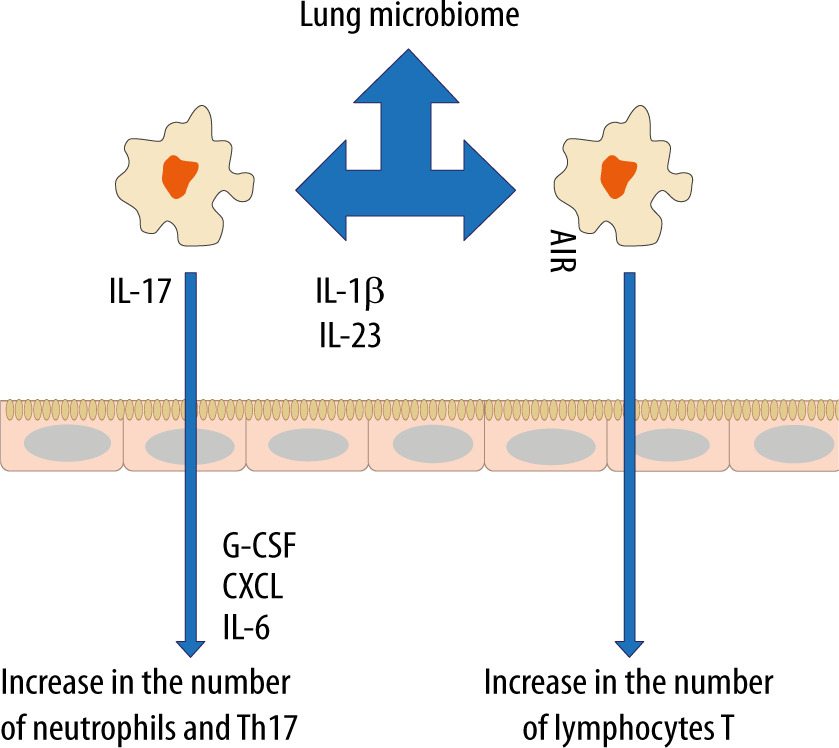
The microbiome also affects the production of antibacterial peptides and proteins (AMPs) in the mucus, which are responsible for inhibiting the multiplication of pathogenic bacteria and for protecting the epithelium [42]. When the lung microbiome is absent or impaired, the production of these proteins is reduced and the susceptibility to infections caused by Pseudomonas aeruginosa, Streptococcus pneumoniae or Klebsiella pneumoniae is increased [43–45]. Of note, in the lungs of animals with the normal microbiome, the number of alveoli was found to be increased [31]. Moreover, it has been revealed that the presence of Proteobacteria increases the risk of developing asthma, partly due to an increased risk of viral infections, especially in the lower airways. Viral infections induce the release of thymic stromal lymphopoietin (TSLP), an epithelial cytokine (alarmin), IL-33 and IL-25 from the airway epithelium, causing type 2 inflammation [46].
THE GUT MICROBIOME AND THE RESPIRATORY SYSTEM
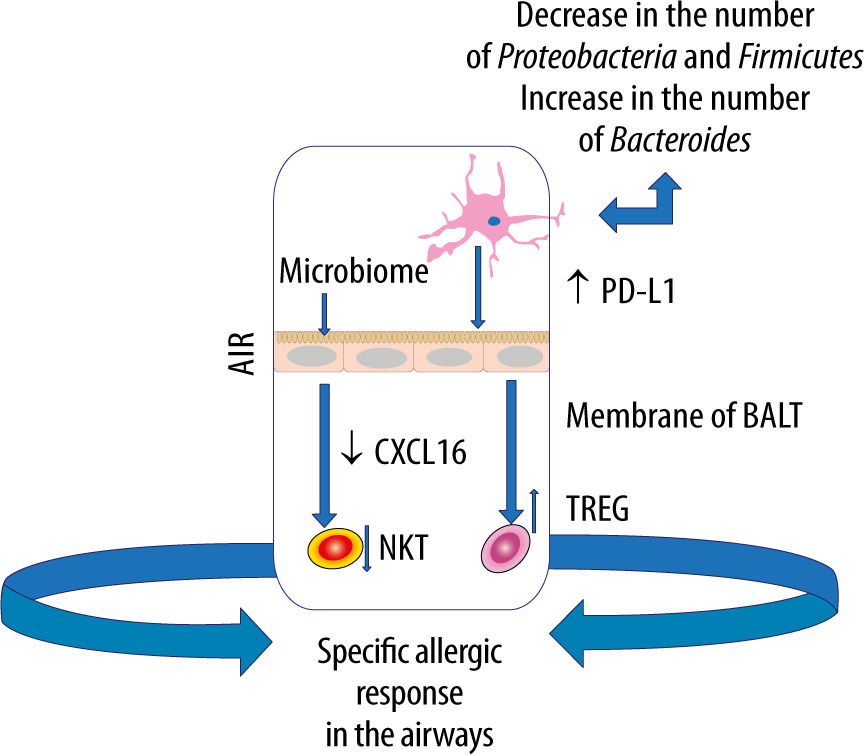
The gut microbiome harbours the largest and most diverse array of commensal bacteria, which shapes the host immune response [47]. It is stressed that imbalance of the gut microflora can affect distant organs, e.g. the brain, liver, skin or heart [48]. Such an imbalance can also contribute to various types of subsequent diseases such as post-traumatic stress disorder in patients with multiple organ injuries [49, 50]. Moreover, a highly significant association has been demonstrated between the gut microbiome and respiratory diseases [21, 51]. It is worth stressing, however, that not only the gut microbiome itself, but also its metabolites can stimulate the immune system within the lungs, forming the gut-lung axis [51, 52]. Metabolites, such as SCFA, have been shown to exert a systemic effect by stimulating the formation of antigen-presenting cells (APCs) filling the airway that induce the type 2 response to a lesser extent [53].
Pro-inflammatory metabolites of the dysbiotic gut microflora may also play an important role in inducing abnormal immune responses in the airways. Experimental studies have shown that the administration of lipopolysaccharide into the bronchial tree causes dysbiosis in the lungs, which leads to disorders of homeostasis within the gut microbiome [54]. Moreover, it has been documented that pneumonia caused by multiple drug resistance (MDR) pathogens, Staphylococcus aureus or Pseudomonas aeruginosa, contributes to endothelial damage and alterations in the gut microbiome [55]. In turn, gastrointestinal dysbiosis, especially in childhood, contributes to the development of asthma [56]. In children with a confirmed risk of asthma, a decreased amount of Rothia, Faecalibacterium, Lachnospira and Veillonella was observed in the GI tract [57]. Experiments in mice have demonstrated that colonization with the above microorganisms and with their metabolites, contributes to a milder course of allergic airway inflammation [58]. Dietary fibre is fermented by commensal bacteria in the colon to SCFAs that have anti-inflammatory effects, maintain gut homeostasis and epithelial integrity, regulate the Treg pool [58–60]. Animals receiving a high-fibre diet have been found to increase the production of SCFAs, including acetate, by the gut microflora, associated with the inhibition of inflammation in the airway. Based on the study results, acetate has been demonstrated to increase the acetylation of forkhead box P3 (FOXP3) by inhibiting histone deacetylase 9 (HDAC9) [61]. This relationship is most likely related to the activation of signallers of acetate-binding GPR 43 as an increased level of acetate in the intestinal loops, e.g. due to the diet used, induces the differentiation of Treg cells and inhibits histone deacetylase [61]. It should be emphasised that the number and functionality of Tregs in asthma is reduced [62]. Subsequent studies have revealed that diets rich in fibre, acting on the dendritic cells and macrophage precursors, stimulate the production of propionate by the microbiome, which reduces the inflammatory response induced by Th2 lymphocytes. In turn, butyrate inhibits the activation of IL-5, IL-13, and group 2 innate lymphoid cells (ILC2s), alleviating the symptoms of allergic pneumonia [63]. Thus, SCFAs produced by bacterial fermentation of dietary fibre modulate oxidative phosphorylation, glycolytic pathways in pulmonary ILC2s and GATA binding protein 3 (GATA3), affecting the production of IL-17a and recruitment of neutrophils into the airway [61–63] (Figure 2).
THE LUNG MICROBIOME IN CRITICALLY ILL PATIENTS
Recently, considerable attention has been devoted to microbiome imbalance in the context of intensive care. In critically ill patients, the gut microbiome and the lung microbiome undergo profound changes. Dysbiosis in critically ill patients has a complex background. In order to explain impaired homeostasis of the lung microbiome, Dickson et al. have suggested three mechanisms influencing the composition and homeostasis of the microbiome [64]. The first is associated with the effects of migration, elimination, and reproduction of commensal microorganisms on the composition of the lung microflora. In this case, the factors determining microbiome reproduction are oxygen pressure, pH, blood flow, alveolar ventilation, temperature, and immune cells [30, 64]. In sepsis and acute respiratory distress syndrome (ARDS), the ongoing inflammatory process changes the physicochemical environment (pH, oxygen pressure, presence of free radicals) and alveolar metabolism. Areas of lung atelectasis and oedema promote an increase in the number of pathogenic microbes and ultimately lead to the development of pneumonia [65]. Furthermore, the oxygen concentration used during therapy has been shown to affect the community of bacteria in the lungs of animals and humans. Recent studies have revealed that hyperoxia causes a selective increase in Staphylococcus aureus in critically ill patients and an altered composition of the microbiome contributes to the development of pneumonia and organ damage [66]. The second mechanism assumes the influence of dietary factors; in healthy individuals, their airways are mainly filled with air and the availability of nutrients for bacteria is relatively limited [67]. Note, however, that in patients with chronic lung diseases, such as cystic fibrosis, chronic bronchitis or asthma, the airways contain dense, protein-rich mucus. Moreover, in ARDS or pneumonia, the alveoli are “flooded” with protein-rich fluid due to damage to the alveolar-capillary barrier, which seems to affect the lung microbiome [68]. The third mechanism is most likely related to intercellular signalling at the molecular level, which can be influenced by glucocorticoids, oestrogens, androgens, neurotransmitters (catecholamine, endogenous opioids) and cytokines such as TNF, IL-1, IL-6 and IL-8 [69, 70]. Imbalance in the lung microbiome is observed in asthma and chronic obstructive pulmonary diseases, as well as in critically ill patients without prior diseases [9, 71, 72] (Figure 3).
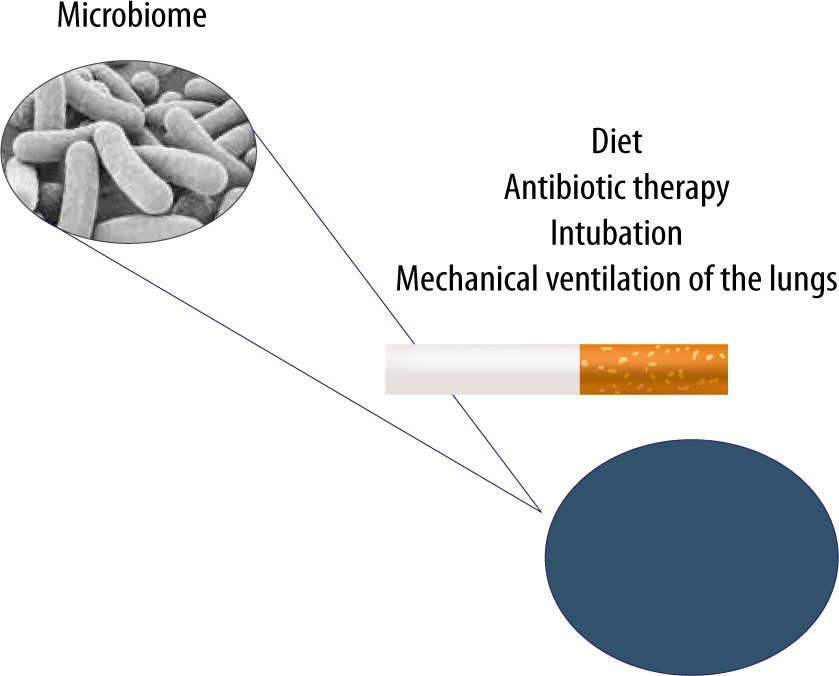
FIGURE 3
The lung microbiome can undergo modifications resulting from improper diet, ongoing infection, antibiotic therapy, cigarette smoking and mechanical ventilation of the lungs in cases of airway pathologies
The lung microbiome imbalance is believed to be affected by antibiotic therapy, endotracheal intubation itself and mechanical ventilation of the lungs [73–75]. The available studies have shown that the lung microbiome is enriched with gut microbes through bacterial translocation facilitated by increased intestinal and alveolar permeability in ARDS and sepsis. The presence of Bacteroidetes and Enterobacteriaceae in the lungs of seriously ill patients, which are characteristic of the gut microbiome, correlates with an increased inflammatory response and may affect the development of ARDS [3, 76, 77]. In cases of increased intestinal and alveolar endothelial permeability, bacteria migrate through the lymphatic system, systemic or portal circulation. The translocation of intestinal bacteria into the lungs is a mechanism potentially affecting the development of lung dysbiosis, inflammation and ultimately lung damage, e.g. associated with mechanical ventilation (called vaping-associated lung injury – VALI) [35]. Early lung dysbiosis in mechanically ventilated patients has been associated with an increase in inflammatory markers – IL-6 and IL-8 – and is strongly related to the development of late ARDS [77, 78]. The observed changes are also responsible for clinically relevant systemic disorders [32, 76]. The presence of artificial airways contributes to continuous micro-aspiration of the oropharyngeal flora and impaired mechanisms of purification of the respiratory tract. It should also be noted that the intubation procedure and mechanical ventilation promote micro-aspiration to the lungs, and the presence of an endotracheal tube significantly impairs the removal of bronchial tree secretions [75, 76]. In critically ill patients undergoing mechanical ventilation, the diversity of bacteria decreases, and opportunistic pathogens can become dominant. Therefore, VALI, considered according to traditional physiopathology, should be modified. Ventilator-associated pneumonia (VAP) should be addressed in terms of the presence of a microbiome and dysbiosis associated with broad-spectrum antibiotic therapy, as well as the need to include molecular techniques to diagnose and accelerate the use of immunomodulatory drugs or probiotics for prevention and treatment [74, 79]. Another important factor influencing the lung microbiome in critically ill patients is antibiotic therapy. Broad-spectrum antibiotic therapy disrupts the homeostasis of the microbiome and affects the development of mechanical ventilation-induced lung injury (VILI) [74, 80]. It is worth noting that impaired homeostasis of the lower airway microbiome is associated with an increased risk of pneumonia [35]. In HIV-infected patients, a close link has been observed between dysbiosis, versus increased Prevotella–Veillonella populations and the risk of severe pneumonia [81]. Moreover, the use of antibiotics, low diversity of the intestinal microbiome and enrichment of the gut microbe with Gammaproteobacteria increase the risk of pulmonary complications in patients after hematopoietic stem-cell transplantation (HSCT) [82]. Commensal microorganisms can also cause pneumonia, as in the case of Staphylococcus epidermidis [83]. This fact seems to prove a close link between the upper and lower airway microbiome and interactions with the population of the entire microbiome [35]. Clinical studies have demonstrated a significant correlation between the severity of ischaemic-reperfusion syndrome, versus impaired homeostasis of the microbiome, activation of the immune system, and epithelial damage [58]. The multiplication of Enterobacteriaceae, accompanied by a persistent inflammatory process, seems of particular importance [84]. Following ischaemic-reperfusion injury, the bacteria characteristic of the intestinal flora are detected in the lungs, serum and mesenteric cells [85, 86]. These bacteria use the activation of TLR 2, TLR 4 and MyD88, inducible nitric oxide synthase (iNOS) and reactive oxygen species to damage the cells. However, it should be stressed that IL-22 signalling and signal transducer and activator of transcription 3 (STAT3) protect the epithelial barrier of the intestines and can prevent translocation of Enterobacteriaceae following burn trauma. Additionally, IL-22 reduces inflammation in the lungs [87]. The impact of nutrition on the microbiome in ICU patients has been an important issue dealt with in many reports over recent years. Diet, quantity and type of individual nutrients affect the species composition of the gut microflora, modulate the number of individual species and their functions as a microbiome [88, 89]. Diets high in animal protein and fats has been found to lead to an increase in the number of Bacteroides in the composition of the gut microbiome, as compared to carbohydrate-high diets, leading to the dominance of Prevotella [90]. Moreover, dietary fibre affects the composition and metabolism of the microbiome. Low fibre intake endangers the mucous layer in the GI tract, causing microbiological instability accompanied by an increase in pathogenic strains and production of potentially harmful metabolites [91]. It should be stressed that in mice with a phenotype associated with high fat content and obesity, an increased number of Firmicutes and Proteobacteria as well as a reduced number of Bacteroidetes have been observed in the microbiome. Weight loss restores the original configuration by limiting fats and carbohydrates [92]. Recent studies have shown that the way nutritional therapy is conducted also diversely affects the microbiome [93]. Total parenteral nutrition changes the gut microbiome by increasing the number of Proteobacteria [94]. In animals receiving total parenteral nutrition, the altered composition of the intestinal lumen has favoured dysbiosis and dysfunction of the epithelial barrier [94]. Parenteral nutrition and fasting are associated with a loss of bacterial diversity that can change the interactions of the microbiome with the host immune system and the ability to control increases in the number of potentially more pathogenic bacteria, e.g. Escherichia coli, Salmonella, Yersinia and Helicobacter, fostering increased expression of pro-inflammatory cytokines in the intestinal mucosa [95]. Such disorders can lead to an increased risk of pneumonia caused by Streptococcus pneumoniae [80]. The mechanisms responsible for the above changes include lower expression of alkaline phosphatase in the enterocytes or activation of TLR receptors leading to the expression of tumour necrosis factor receptor (TNFR) in the epithelial cells, lower expression of cytoskeleton proteins and increased bacterial translocation [96]. Moreover, animals receiving total parenteral nutrition have demonstrated increased expression of interferon-γ in the intestinal epithelium [97]. On the other hand, enteral nutrition has been found to exert anti-inflammatory effects, as reflected in decreased concentrations of pro-inflammatory cytokines, TNF-α and IL-6, in serum and increased concentrations of anti-inflammatory cytokine IL-10, which is associated with lower mortality rates [93, 98].
THERAPEUTIC DIRECTIONS
Numerous studies have demonstrated that maintaining the microbiome balance can be another important therapeutic direction in critically ill patients treated in intensive care units. Intestinal microflora-targeted dietary interventions can potentially improve clinical outcomes, taking into account the connection between the GI tract and the respiratory system. In medical practice, the effects of nutrients on the microflora are neglected. Therefore, the purpose of nutritional therapy, especially in critically ill patients, should be broadened. Fibre-rich diets alter the microbiome in the GI tract or respiratory system and increase the concentration of SCFAs in the blood, reducing the inflammatory process associated with allergies and mortality rates in lung diseases [14, 99–101], as dietary fibre is a product consumed by commensal bacteria for the biosynthesis of SCFAs [102]. In patients with asthma, the diet has been shown to affect the systemic inflammatory response and the diet rich in fruit and vegetables exerts positive effects on the ongoing diseases [103, 104]. Analysis of the reports available in literature has revealed that the use of probiotics in ICUs reduces the incidence of ventilator-associated pneumonia without affecting the mortality and length of hospitalization [105]. Supplementation with probiotic bacteria such as Lactobacillus rhamnosus, Bifidobacterium lactis and B. breve, affects the inflammatory response to allergic lung diseases [106, 107]. Moreover, the use of Lactobacillus rhamnosus and Bifidobacterium breve in smokers with COPD inhibits the release of pro-inflammatory mediators by macrophages in response to cigarette smoke [108]. Moreover, it should be highlighted that probiotics have immunomodulatory effects, stimulating the natural smoking-suppressed activity of NK cells [109]. When analysing the treatment administered in critically ill patients, the effect of antibiotic therapy and steroid therapy on the lung microbiome should be considered. Inadequate antibiotic therapy in patients treated for COPD exacerbations has significantly impaired homeostasis of the lung microbiome [110–112]. Furthermore, it has been documented that the antibiotics reduce the number and diversity of microflora, while steroid therapy increases the number of Moraxellaceae, Pasteurellaceae, Pseudomonadaceae and Enterobacteriaceae [113]. Combined steroid and antibiotic therapies have resulted in a significant increase in Proteobacteria in the lungs [114]. However, the above disorders seem to occur only in critically ill patients, as in patients with mild to moderate asthma, the Proteobacteria family dominates, suggesting that dysbiosis is independent of steroid therapy [113]. Nevertheless, the observations in question come from the studies carried out in a small population of patients and have to be confirmed in multicentre studies.
CONCLUSIONS
The lung microbiome plays an important role in lung diseases. The interactions between impaired lung and gut microbiomes seem to be essential for the management of patients in intensive care units. Demonstrating such interactions should also elucidate the mechanism of the gut–lung axis and suggest a new therapeutic direction, fundamentally changing the treatment of critically ill patients.