

 POLSKI
POLSKI







Introduction
The programs funded by the National Health Fund (NFZ) in Poland define the criteria for the inclusion of short-statured children with growth hormone deficiency (GHD), born small for gestational age (SGA), with Turner syndrome (TS), Prader-Willi syndrome (PWS), and chronic kidney failure (CKF) into the treatment with human recombinant growth hormone (rhGH). Therapeutic programs specify individually for each diagnosis the principles of diagnostic management, criteria of inclusion into therapy, the dosage of rhGH, monitoring procedures of the therapy, and the exclusion criteria. The aim of the study is to develop indications for genetic diagnostic management of children treated with rhGH diagnosed with SNP and SGA, in cases of insufficient improvement in growth during treatment with rhGH or when coexistence of other disorders reduces the effectiveness of treatment.
Materials and methods
Four boys were analyzed: three patients diagnosed with SNP at the ages of: 8 years and 2 months – patient 1, 13 years and 2 months – patient 2, 16 years and 6 months – patient 3, and patient 4 diagnosed with SGA at the age of 6 years and 11 months. Growth curves of all the boys were below the 3rd percentile on our national growth charts [1]. Idiopathic and structural causes of rhGH deficiency, absorption disorders and other concomitant diseases that could have impaired their growth were excluded.
Hormonal tests
Growth hormone (GH) deficiency was diagnosed by showing GH concentration < 10 ng/ml in 3 stimulation tests: 2 pharmacological tests (after the administration of clonidine, glucagon, or arginine) and an overnight test. In recent years, 2 pharmacological tests have been used, performed in accordance with the guidelines included in the Polish recommendations regarding therapy with rhGH accepted by the Polish Ministry of Health. Hormonal assays of growth hormone (GH), luteinizing hormone (LH), follicle-stimulating hormone (FSH), testosterone, adrenocorticotropic hormone (ACTH), cortisol using the radioimmunoassay method on a Siemens Centaur XPT analyzer were performed. The insulin-like growth factor (IGF1) levels with values depending on age and gender were determined using the immunochemical method with a radioactive label (Dia Source).
Genetic tests
Genetic tests were performed using various genetics methods, i.e. whole exome sequencing, Sanger sequencing, array CGH, MLPA, karyotype analysis. For molecular methods, DNA was prepared from peripheral blood using standard procedures and obtained after informed consent. The array CGH (aCGH) analysis was performed using the Agilent array ISCA 8x60K V2 according to the manufacturer’s protocol. Data analysis was performed using the Agilent Cytogenomics software. Oligo positions were referred to the UCSC Genome Browser (hg19). MLPA (Multiplex Ligation-dependent Probe Amplification) was performed according to the manufacturer’s protocol. The patient was diagnosed using the SALSA MLPA kit P018-F1 SHOX. The whole exome sequencing (WES) and the analysis of the FGFR1 gene using the next generation sequencing (NGS) were performed according to the manufacturer’s protocol. Sanger sequencing was performed to analyze the GLA gene and confirm variants of interest identified by the NGS method. For cytogenetic karyotype analysis, peripheral blood leukocytes were isolated from a heparinized blood sample, cultured, prior to evaluation by high-resolution G-banding of at least 30 metaphase nuclei.
Neuroimaging tests
Magnetic resonance imaging (MRI) of the pituitary-hypothalamic region with gadolinium contrast was performed in all the patients in the AP and sagittal projections, and the images in T1 and T2 projections were analyzed. The MRI examination was performed using GE HDX Echospeed 81.5 T and GE Optima 450 W, 1.5 T units. In all the boys, magnetic resonance imaging excluded pituitary abnormalities. After considering the conclusions, rhGH treatment was initiated at appropriate doses. Treatment was monitored according to the recommendations of the NFZ (National Health Fund) therapeutic programs for children with SNP and SGA.
Results
All the boys showed a lack of the expected improvement in growth. The growing process in the boys before and after the initiation of treatment are presented in Table I.
Table I
Growth velocity of patients before and after initiating treatment
Patient 1
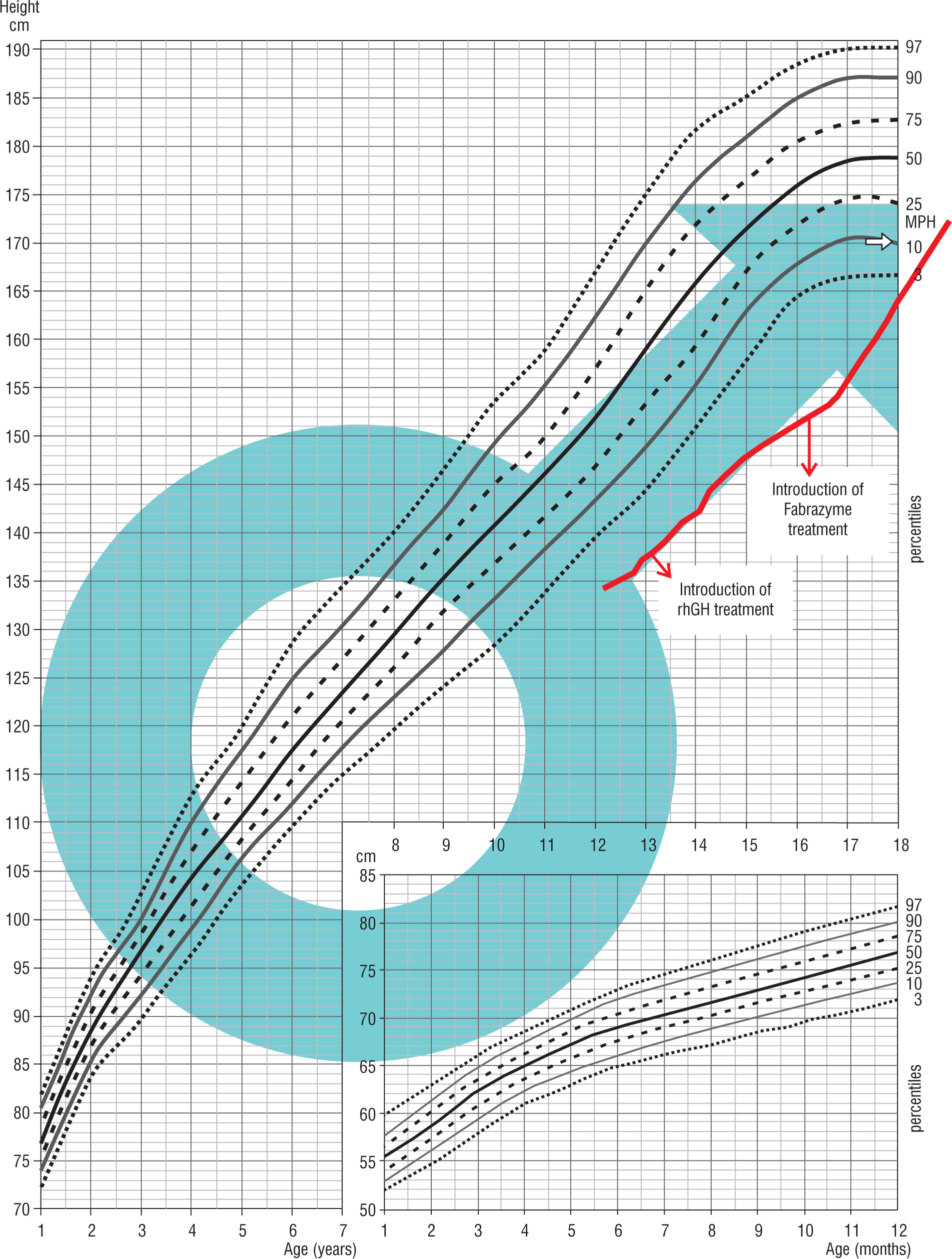
A child born on January 1, 2013, at 40 weeks of gestation, with the birth weight of 3.3 kg, the birth length of 54 cm, and the head circumference of 33 cm. He was born from the first pregnancy terminated by a cesarean section due to the lack of progress in labor and a threat of fetal distress. His Apgar score was 4/5/8 points at 1/3/5 minute after delivery. Prenatal ultrasound did not reveal a heart defect in the child. After birth, coarctation of the aorta was diagnosed, which was corrected at 19 months of age (August, 2014). The hormonal diagnosis of growth deficiency was initiated at the age of 5 years. Until the beginning of the diagnostic process, the boy’s growth was below the 3rd percentile. In his family history, the mother was diagnosed with Hashimoto’s disease, Raynaud’s disease. The mother’s height was 158 cm and the father’s height was 173 cm, the mid-parental height (MPH) was assessed as 173 cm – between the 25th and 10th percentile. Physical examination revealed some dysmorphic features: a prominent fontanelle, mildly slanting palpebral fissures, widely spaced nipples, shortened right lower limb, hypoplastic nails. At the age of 8 years (February, 2021), SNP was diagnosed (max. GH release in the arginine test was 1.74 ng/ml, in the glucagon test it was 2.69 ng/ml). The treatment with rhGH was started at the age of 8 years and 2/12 (February 2022). After the initiation of the treatment, nocturnal vomiting episodes occurred, which recurred after each attempt at increasing the rhGH dose. After reducing the dose of rhGH, the episodes of headache with vomiting occurring for up to 3–4 hours appeared less frequently, about once a month. Genetic and neurological diagnostic management was ordered. In August 2021 the result of genetic consultation was: karyotype from blood lymphocytes: 46, XY [16] / 45, X [14] – abnormal male karyotype – mosaic karyotype: monosomy of chromosome X occurs in about 50% of analyzed metaphases. Due to vomiting precluding effective rhGH dosing and no improvement in growth, the rhGH treatment was discontinued in April 2022 and neurologic consultation was ordered. In October 2022, in spite of normal results of awake and sleep electroencephalograms, due to the overall clinical picture, a prophylactic treatment was initiated with valproic acid. Suspicion of Panayiotopoulos syndrome was raised. In December 2022, resumption of rhGH treatment at a higher dose was started and no vomiting observed (Fig. 1).

Patient 2
Patient 2 was born on September 22, 2002 by a cesarean section, after an induction of the labor, from the head-first delivery at 39 weeks of gestation, with the birth weight of 3.95 kg, the birth length of 56 cm, and the head circumference of 35 cm; his Apgar score was 10 at 1 minute after delivery. His medical history included an intussusception treated with an enema (December 2002), and an orchidopexy due to right-side cryptorchidism (January 2013). The child occasionally reported burning sensations in the hands and feet. The family history, except for migraines in his mother, was unremarkable. The mother’s height was 162 cm, the father’s height was 176 cm, the mid-parental height (MPH) was assessed as 175.5 cm – 25th percentile. The hormonal diagnosis of the patient’s short stature was initiated at the age of 11. Until the start of the diagnostic process, the boy’s growth was below the 3rd percentile. The boy was diagnosed due to growth deficiency. On December 2015, SNP was diagnosed (max. GH release in the glucagon test was 4.29 ng/ml, in the clonidine test was 0.47 ng/ml, in the overnight test was 7.08 ng/ml). At the age of 13 years and 2 months, treatment with rhGH was initiated. Despite increasing rhGH doses, the expected improvement in the growth rate was not achieved; moreover starting from autumn 2017, burning sensations in the hands and feet intensified. In September 2017, the boy was brought to the emergency room with fever, burning sensations in the hands and feet, and difficulty in movement. In November 2017, rhGH treatment was discontinued, and the child was referred for rheumatology diagnostics. In November 2017, enzymatic examination for Fabry’s disease confirmed the α-galactosidase deficiency. In October 2018, a genetic opinion confirmed Fabry’s disease with a hemizygous mutation c.109 G>C in the GLA gene. In February 2019, enzymatic replacement therapy with α-galactosidase A (Fabrazyme) was initiated, which also did not improve the child’s growth. In May 2019, the rhGH treatment was reintroduced with an improvement of his growth rate (Fig. 2).
Patient 3
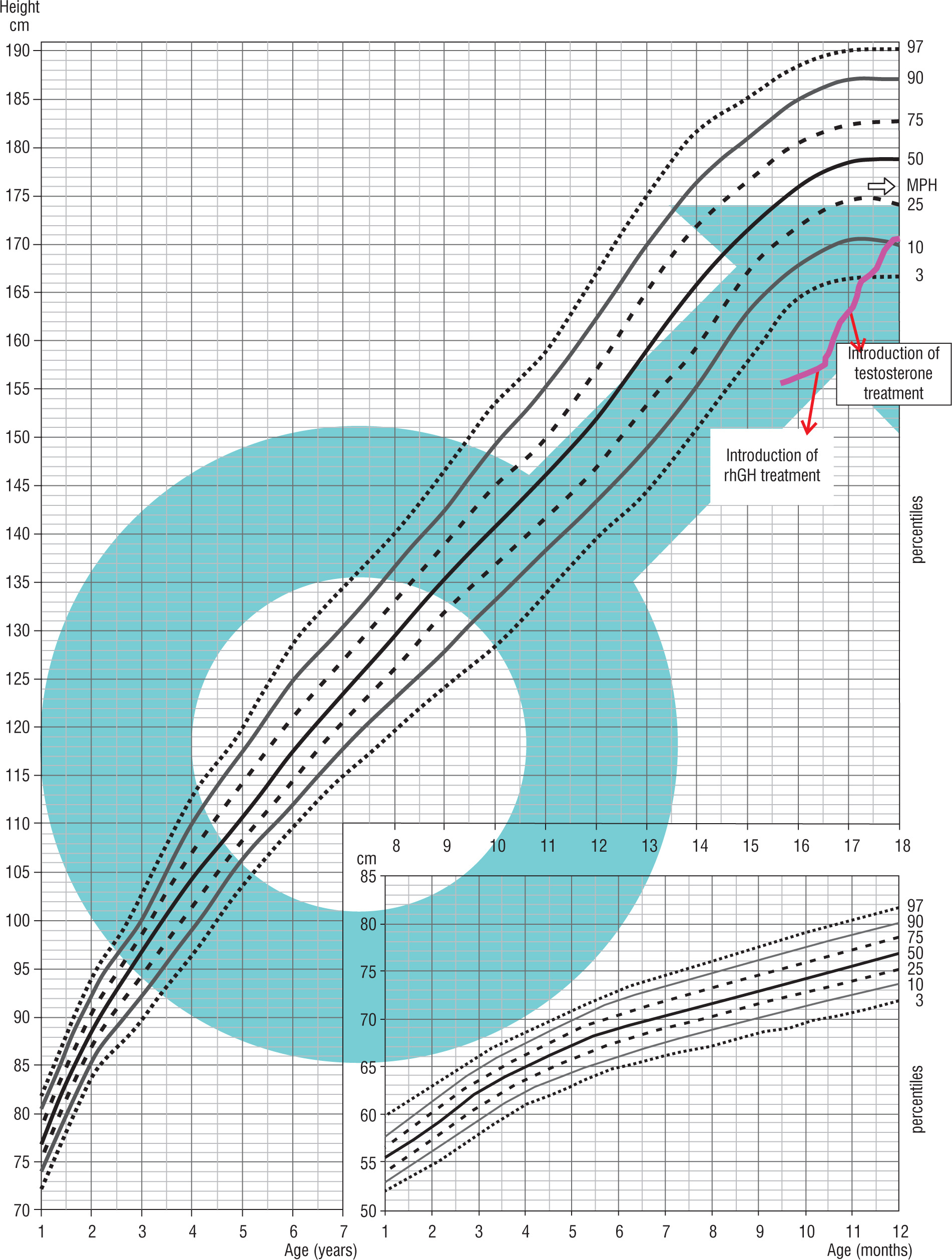
Patient 3 was born on October 22, 2004, from the first pregnancy and the head-first delivery at 39 weeks of gestation, with the birth weight of 3.95 kg, the birth length of 53 cm, the head circumference of 34 cm, and the chest circumference of 33 cm, His Apgar score was 8/8/9/9 in 1/3/5 minute after delivery. His medical history included a bilateral orchidopexy (August 2019, and February 2020). The child remained under gastroenterological care due to gastric mucosa inflammation. The family history, except for asthma in his father, was unremarkable. His parental heights were: mother – 169 cm, father – 171 cm, MPH amounted to 176 cm – 25th percentile. The hormonal diagnosis of growth deficiency was suggested when he was 15 years of age. His medical history indicated a slowdown in growth from approximately age of 13 years. Physical examination revealed some abnormalities: linear scars after abdominal surgery on both sides, a longitudinal café-au-lait spot about 3 cm above the left iliac crest, slight knee valgus, syndactyly of the 3rd and 4th toes on the right foot, micropenis. On July 2020, an MRI examination of the hypothalamic-pituitary area revealed a slightly concave upper outline of the pituitary gland, and the signal of the anterior lobe of the pituitary was not entirely homogeneous. Due to the MRI result and the patient’s lack of the sense of smell, a radiological-endocrinological consultation was held on September 8, 2020, to verify the presence of olfactory bulbs in the MRI examination of the hypothalamic-pituitary area. The patient was referred for a genetic diagnostic management. On March 2021, SNP was diagnosed (max. GH release in the glucagon test was 5.62 ng/ml, in the clonidine test it was 5.32 ng/ml, in the night test it was 2.32 ng/ml). At the age of 16 years and 5 months, rhGH treatment was initiated, and at the age of 17 years, due to prepubertal LH-RH test results and low testosterone levels (October 2021), testosterone treatment was started. On February 2022, the result of the NGS panel gene analysis associated with hypogonadotropic hypogonadism showed a heterozygous variant c.2198 T>C in the FGFR1 gene, as a confirmation of FGFR1-dependent Kallmann syndrome (Fig. 3).
Patient 4
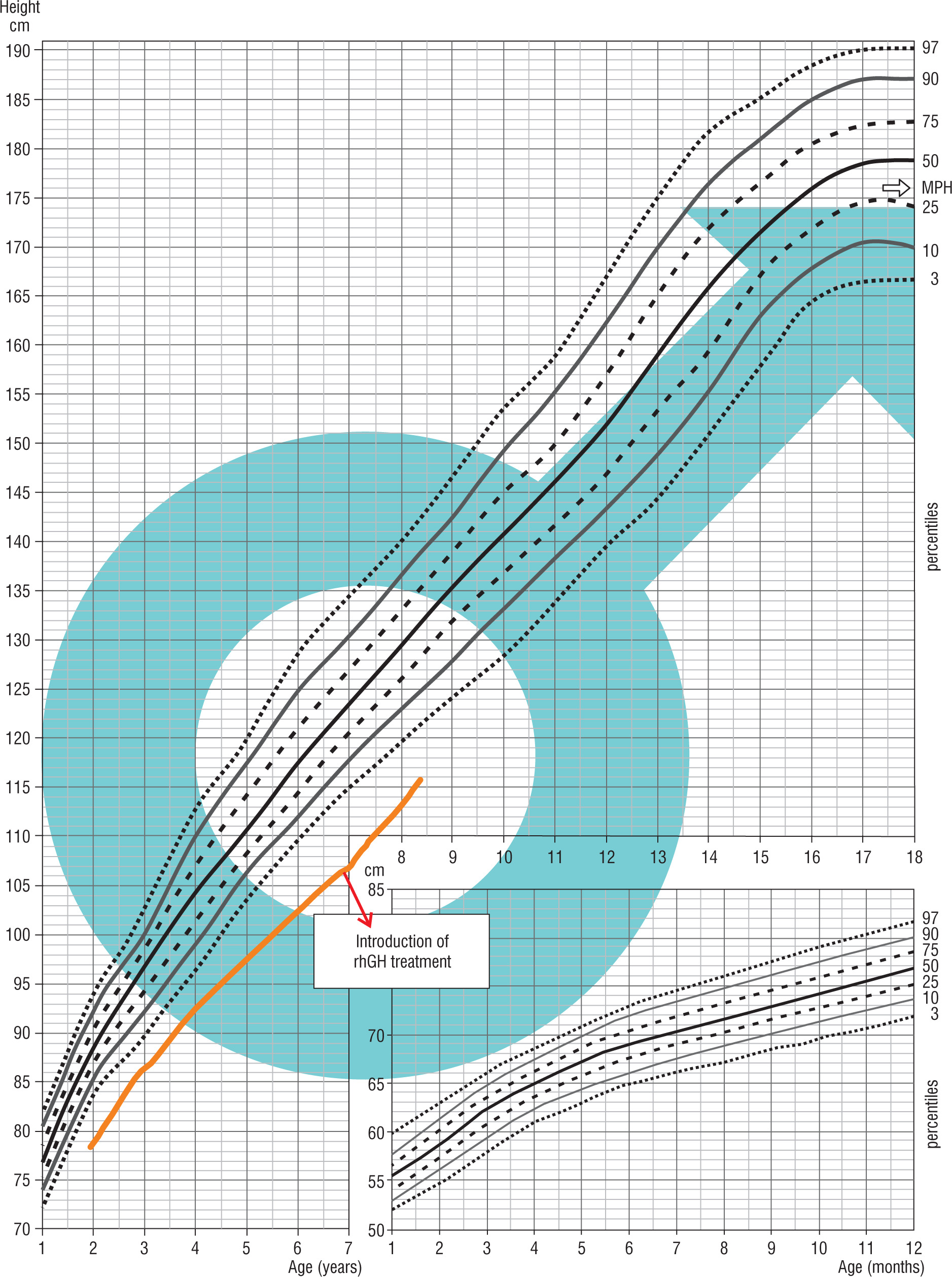
Patient 4 was born on May 30, 2014, from a second pregnancy and second delivery terminated by a cesarean section due to orthopedic indications in the mother and a breech presentation of the fetus, after 39 weeks of gestation, with the birth weight of 2,770 g (–2.15 SDS), the birth length at birth of 50 cm, the head circumference of 34 cm and the chest circumference of 32 cm. His Apgar score was 10 points at 1 minute after delivery. Fetal ultrasound examinations revealed increased nuchal translucency and hypoplastic nasal bone. In the PAPP-A (pregnancy-associated plasma protein-A) test, an elevated β-hCG level was observed, leading to chorionic villus sampling with a karyotype result of 46XY. His family history was unremarkable. His parental heights were: mother – 164 cm, father – 175 cm, MPH amounted to 176 cm. The hormonal diagnosis of his short stature was initiated at the age of 3 years. Until the start of the diagnostic process, the boy’s growth was below the 3rd percentile. Some abnormalities were found in his physical examination and included coarse facial features, hypertelorism, wider nasal base, rounded nasal tip, deep nasolabial furrow, depressions in the soft tissues below the eyes, and puffy dorsa of the feet. Due to dysmorphism and the short stature, the patient underwent genetic and endocrinological diagnosis. In 2016, the results of aCGH were normal, in 2017, the organic acid profile in urine using GC/MS was normal, and in 2020, the Face2Gene program suggested a clinical diagnosis of KBG syndrome based on facial images. In 2021, the hormonal diagnostic tests were completed, showing a maximum GH release in the glucagon test of 20.48 ng/ml and in the overnight test the value was 6.24 ng/ml. The child was diagnosed with SGA, and treatment with rhGH was initiated at the age of 6 years and 11 months. In February 2022, during the WES analysis, a heterozygous de novo variant c.2728_2729delAG in the ANKRD11 gene was detected, correlating with KBG syndrome (Fig. 4).
Discussion
In all girls with growth deficiency, Turner syndrome should be ruled out, while in boys, the inclusion criteria for therapeutic programs for SNP and SGA do not necessitate genetic testing. Nevertheless, the presented cases suggest that genetic testing is essential in the presence of dysmorphic features, congenital abnormalities, or other irregularities. In Patient No. 1 the presence of aortic coarctation was a sufficient reason for genetic testing, which confirmed the mosaic Turner syndrome. With neurological diagnosis and the diagnosis of epilepsy, effective treatment was possible. Lack of a proper response to rhGH treatment and the presence of atypical symptoms (burning sensations in the hands and feet) in patient No. 2 prompted genetic testing which revealed the ultra-rare mucopolysaccharidosis – Fabry’s disease, affecting 6–8% of the population in Poland. Enzymatic treatment alone did not improve the growth rate in this child; only the combined treatment with enzymatic replacement therapy with α-galactosidase A and rhGH did prove effective. In patient No. 3, delayed puberty prompted testing to rule out multihormonal pituitary deficiency. An interview indicating olfactory disorders and MRI results suggested the necessity of genetic testing, which confirmed Kallmann syndrome. The estimated frequency of this syndrome is usually 1 : 10,000 [2]. Classical Kallmann syndrome (HH1) is inherited in an autosomal dominant manner, linked to the X chromosome – a defect in the Xp22.3 region of the KAL1 gene [2]. Autosomal recessive inheritance has also been confirmed, and mutations in the genes correlated with this syndrome have been identified (e.g., FGFR1, PROK2, PROKR2, GNRHR) [3–6]. Low concentrations of GH in stimulation tests were observed in the boy, possibly due to his delayed puberty and lack of priming. However, hypotheses suggest that mutations in genes underlying Kallmann syndrome may also lead to pituitary insufficiency. These opinions are based on the knowledge that the embryonic structure called the preplacodal region gives rise to the olfactory, lens, and pituitary placodes. Therefore, defects in the genes involved in the development of these structures may be responsible for both phenotypes [7]. So far, many genes previously associated with hypogonadotropic hypogonadism have also been linked to IGHD or CPHD with or without complex phenotypes. There is a significant genetic heterogeneity with autosomal dominant or recessive inheritance with incomplete penetrance. Among the described clinical pictures are various combinations of pituitary hormone deficiencies associated with SOD, HPE, PSIS, and Moebius syndrome [7]. The boy was treated with rhGH and testosterone with positive results. In Patient No. 4 who was small for gestational age (SGA) and presented with dysmorphism, genetic tests revealed the KBG syndrome. The KBG syndrome is a rare disorder characterized by intellectual disability and associated with macrodontia of the upper central incisors, specific craniofacial findings, short stature and skeletal anomalies. Genetic corroboration of a clinical diagnosis has been possible since 2011, upon identification of heterozygous mutations in or a deletion of the ANKRD11 gene [8–10]. The frequency of growth hormone deficiency-induced short stature ranges from 1 in 4,000 to 1 in 10,000 live births, and 3–30% of subjects have a genetic basis [11]. Genetic forms of GHD can occur as isolated GHD (IGHD) or as part of combined pituitary hormone deficiency (CPHD). The IGHD can be sporadic or familial with various types of inheritance: autosomal recessive (IGHD IA and IB), autosomal dominant (IGHD II), and X-linked (IGHD III) [6, 10]. The GH1 gene, encoding growth hormone (GH), is located in the chromosomal region 17q23.3 and consists of five exons. Mutations in GH1 result in IGHD; however, some types, especially IGHD II, may manifest as CPHD [11]. The GH1 gene deletions were identified as the first monogenic cause of GH deficiency. Recessive autosomal mutations in GHR were later discovered, causing complete GH insensitivity and severe growth deficiency [11]. In the 1990s, mutations in genes encoding transcription factors and signaling molecules involved in pituitary ontogenesis and development were identified. The analysis of spontaneous mutations and transgenic mouse models demonstrated the role of many of these factors, including HESX1, PROP1, PIT1, LHX3, LHX4, SOX2, SOX3, OTX2, PAX6, FGFR1, SHH, GLI2, and FGF8, in the etiology of congenital pituitary insufficiency. Such insufficiency can feature a deficit in one or more pituitary hormones. The phenotype can be highly variable, with different gene mutations inducing the same phenotype [7, 12]. Abnormal function of genes regulating the differentiation of Rathke’s pouch and the development of its endocrine properties result in disorders affecting both the pituitary function and morphology. Developmental defects most frequently associated with CPHD include adenohypophysis agenesis, septo-optic dysplasia (SOD), congenital Rathke’s cleft cysts, and holoprosencephaly [7, 12]. Genetic analysis should be performed in children exhibiting malformations associated with the risk of congenital pituitary insufficiency, such as isolated aplasia or hypoplasia of the pituitary gland, hypoplasia of the anterior pituitary lobe and/or ectopia of the posterior lobe, pituitary stalk agenesis, or empty sella syndrome [12]. The advancements in high-throughput sequencing techniques have unveiled a spectrum of over 40 genes that affect the growth hormone (GH) secretion, bioavailability, and insulin-like growth factor-1 (IGF-1) function [7]. Notably, the newly discovered genes (GLI2, HPE, FGFR1, PAX6, GLI3, ARNT2, CDON, GPR161, IGSF1, PROKR2, TGIF1, TCF7L1, PROK2) have been associated with growth hormone deficiency, either in an isolated form (IGHD) or in combination (CPHD) [7, 13]. These genes are linked to various clinical features, including polydactyly, SOD (short stature, optic nerve atrophy, and Pelger-Huët anomaly), Moebius syndrome, craniofacial abnormalities, optic nerve disorders, Hirschsprung’s disease, and macrorchism. Additionally, novel genetic defects within the GH-IGF axis (STAT5B, STAT3, PAPPA2, PIK3R1, IGF2) serve as indications for broadening the diagnostic criteria for short stature in children who are born small for gestational age (SGA) [13]. This expansion is particularly relevant for cases with abnormal proportions, extreme short stature, microcephaly, immunological disorders, and dysmorphic features. Some presentations may resemble the Silver-Russel syndrome phenotype, characterized by low concentrations of IGF2. These genetic insights contribute to a more comprehensive understanding of the genetic underpinnings of short stature in diverse clinical contexts. The authors of the 2023 consensus for SGA children emphasize the importance of genetic testing in children born with SGA who have such features as skeletal dysplasia, GH-IGF1 axis defects, dysmorphic features, microcephaly, intellectual disability or developmental abnormalities that may suggest syndromic growth deficiency. The afore-mentioned authors present a detailed analysis of mutations leading to development of small-for-gestational-age (SGA) children with normal and reduced head circumference, imprinting disorders and methylation disturbances, as well as pubertal disorders and advanced bone age [13]. They also emphasize the need for genetic testing in children born SGA with persistent short stature, with or without skeletal disproportion, and for whom no other cause of such deformities is known. Children with syndromic SGA are usually shorter before treatment is initiated and require higher doses of rhGH to achieve the height gain as compared to patients with SGA without genetic abnormalities [14]. The age of initiation of treatment, the degree of growth deficiency and the appropriate dosage of rhGH also play a role in the effects of treatment [15]. According to the 2023 Consensus, a Small for Gestational Age (SGA) child is defined as having a birth weight and/or length below –2 SDS for gestational age. Catch-up growth is defined as a growth rate (cm/year) greater than 0 SDS, meaning above the median for chronological age and gender. Adequate catch-up growth is defined as growing into the normal range, taking into account the mid-parental height. Short stature is defined as growth below –2.0 SDS for age and gender. The terms intrauterine growth restriction (IUGR) and SGA are often used interchangeably, but they should not be used synonymously. IUGR signifies an abnormal increase in estimated fetal weight and abdominal circumference during a specific period of pregnancy based on two ultrasound measurements, irrespectively of the size at birth [13].
Conclusions
The presence of dysmorphic features and symptoms atypical for growth hormone deficiencies could warrant genetic diagnostic management before initiating treatment.
Lack of significant improvement in growth is an indication for reevaluation of patients who have not completed growth.
Genetic studies in this patient group often explain the causes of a slow growth rate.
Re-evaluation of recommendations for treatment of children with SNP and SGA treated with rhGH is needed, especially in the era of new, modern genetic diagnostic procedures and higher possibility to precisely diagnose genetic disorders associated with short statute.
The present authors have developed a proposal for a multicenter program aimed at establishing indications for genetic diagnosis in children diagnosed with SNP and SGA and treated with rhGH.
We propose the following inclusion criteria for genetic analysis in children from the NFZ programs for small for gestational age (SGA) and growth hormone deficiency (GHD). The program has been approved by the Bioethics Committee.
Disproportionate body structure, dysmorphism, bone anomalies.
Intellectual disability.
Congenital developmental defects (heart defects, kidney defects, central nervous system defects, and others).
Disorders/lack of puberty/lack of pubertal growth spurt.
Autoimmune, storage and systemic diseases.
Multihormonal pituitary deficiency.
Elevated or significantly decreased IGF1 levels in patients with slow growth.
Suspicion of fetal alcohol syndrome (FAS).
Lack of improvement in the growth rate after initiation of rhGH as compared to growth rate before treatment – < a twofold increase in the growth rate in the first year of treatment or height velocity delta < 0.5 SDS.
Height gain during rhGH treatment < 4 cm/year before reaching skeletal maturity, above 14 years for girls and above 16 years for boys in the second and subsequent years of treatment.
We propose the following exclusion criteria from genetic analysis:
Manifestation or relapse of an oncological disorder.
Lack of cooperation with parents.
Lack of consent for genetic testing.
Moreover, based on our observations and literature reports, we propose a multicenter collaboration to improve the diagnosis of short-statured children with growth hormone deficiency (GHD) and small for gestational age (SGA).