

 ENGLISH
ENGLISH







Introduction
Lysosomal storage diseases (LSDs) are inherited metabolic disorder diseases; these kinds of diseases are caused by enzyme deficiencies resulting in accumulation of un-degraded substrate. This storage process leads to clinical manifestations depending on the specific substrate and site of accumulation. LSDs are a heterogeneous group of diseases [1]. They are characterized by accumulation of macromolecules in lysosomes, like glycosaminoglycan (GAG). If GAG degradation cannot be performed normally, the resulting LSD is called mucopolysaccharidosis (MPS). Here the degradation of carbohydrate complex is hampered and thus, GAG cannot be processed normally [2]. Mucopolysaccharidoses constitute ~30% of all LSDs [3].
Mucopolysaccharidosis
Mucopolysaccharidoses are known as a group of metabolic diseases caused by absence or malfunction of an enzyme being necessary for breakdown of molecules long chains (carbohydrates) in the cells. In such patients deficiency of 11 different enzymes being required for breakdown of carbohydrates in small and simple molecules have been reported. These GAGs are accumulated in blood, brain, spinal corda and different connective tissue and are there responsible for local cell damage. Symptoms may be similar or may vary among different types of MPSs [4].
The prevalence of MPSs is different in different countries and varies between 0.07 in Australia and 4.05 per 100,000 life-births in Estonia; reported differences may be due to undiagnosed cases and/or different genetic prevalence: It must be suggested that MPSs are underdiagnosed in many countries due to lack of corresponding diagnostic means.
Mucopolysaccharidoses are known to be autosomal recessive disorder, meaning that only individuals with two mutated alleles are affected. When both parents are heterozygote for the disease causing allele in each pregnancy there is a one in four chance that the child will be affected. Accordingly, unaffected persons in a family may be either heterozygous for a disease causing allele, or may carry two non-disease causing ones. Only MPSII or Hunter syndrome is an X-linked recessive disorder [5–7].
Generally, following factors may increase the chance to have or pass an MPS-disease-causing allele:
family history of an MPS;
a couple is consanguineous or derive from same distinct ethnic or geographically clustered community;
a couple is heterozygous for MPS disease causing allels.
Among all types of MPS, MPS III (or Sanifilippo syndrome) is most frequent; it was first described ~50 years ago [8]. Here heparin sulfate (HS) plays a pivotal role, as it does also in neuronal development [9]. Accordingly, neurological pathology found in MPS III patients can be explained. Relevance of lysosomes in maintaining of neuron and brain homeostasis has been reported recently [10]. Impairment of HS metabolism, where also lysosomes are involved and accumulation of α-synuclein, connected MPS III with other neurological diseases such as Parkinson disease is reported [11]. On the other hand MPS III can be seen in one line with autism spectrum disorders (ASDs) which are associate with neurodevelopmental delay, including language, impaired social interactions and restrictive, repetitive and stereotyped behaviors [12, 13].
Mucopolysaccharidosis III can be divided into five subtypes [3]. These are caused by mutation of sulfamidase (MPS IIIA; OMIM#252900) [14], α-n-acetylglucosaminidase (NAGLU, MPS IIIB; OMIM#252920) [15], heparan acetyl CoA: α-glucosaminide N-acetyltransferase (HGSNAT, MPS IIIC; OMIM#252930) [16], and N-acetylglucosamine 6-sulfatase (GNS, MPS IIID; OMIM#252940) [17].
Here we describe the molecular basis of MPS III, and summarize therapeutic approaches. The process of heparin sulfate degradation and enzymes which are involved in this process are presented in Fig. 1.
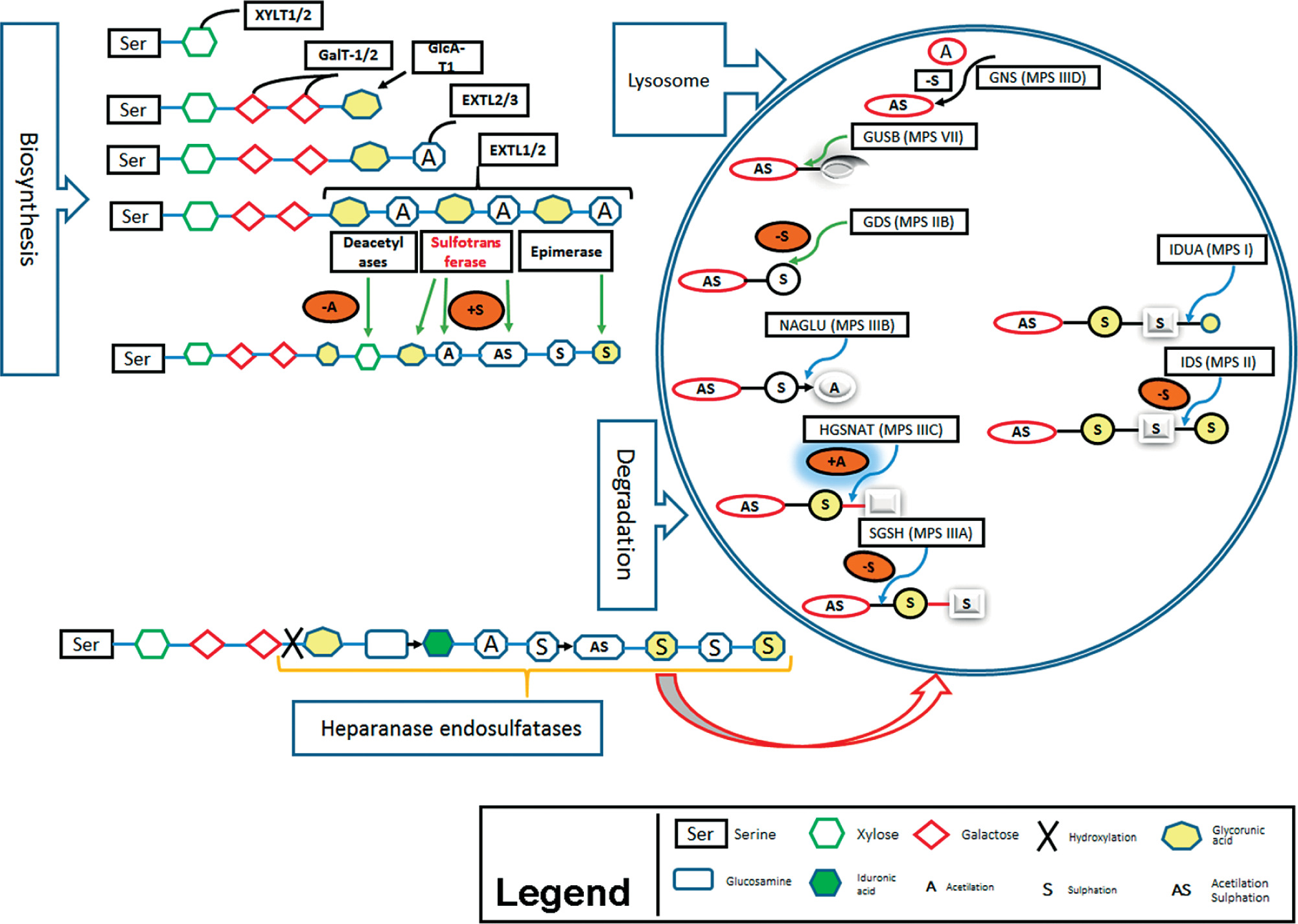
Genetics of mucopolysaccharidosis III
The known common mutations being responsible for MPS III are presented in Table IIa. Besides, other recently found genes involved in MPS III in Table IIb (only single case reports, yet). Approximate frequencies of genes involved in MPS IIIA, MPS IIIB, MPS IIIC, MPS IIID and MPS IIIE were determined based on reports in Varsome and ClinVar databases [99–100].
Mucopolysaccharidosis IIIA
Mucpolysaccharidosis IIIA (~27% of MPS III) is caused by mutations in the gene SGSH localized in chromosome 17q25.3 (Table II, III). This gene is responsible for coding of sulfaminidase (heparin sulfate sulfatase or N-sulfoglucosamine sulfohydrolase) [18]. The first patients diagnosed with symptoms of MPS IIIA were described by Sanfilippo et al. [8].
Specific mutations in sulfaminidase are found in variant frequencies in different countries. The missense mutations p.R74C occurs at a frequency of 56% in Polish MPS III population [19]. In Australia amino acid change at p.R245H is most frequent with 31%; in Germany frequency of this mutation is 35%, and in Netherlands 58% [19, 20]. In Italian patients missense mutation for p.S66W was found with a frequency of 29% [21].
Table I
Incidences of MPS in different ethnicities
Mucopolysaccharidosis IIIB
Mutation in gene NAGLU (17q21.2) causes MPS IIIB (~36% of MPS III; Table II). This gene encodes N-acetyl-α-glucosaminidase, a lysosomal enzyme of 720 amino acids with six glucosylation site [22]. Most frequently observed are missense mutations p.F48L, p.G69S, p.S612G and p.R643C which are associated with late-onset phenotype [23–25].
Table II
Summary of the phenotypic, enzymatic, and genetic classification of the MPS III
Mucopolysaccharidosis IIIC
The third form of mucpolysaccharidosis, MPS IIIC (~30% of MPS III), is caused by lysosomal membrane acetyl-CoA α-glucosaminide N-acetyltransferase coded by HGSNAT gene (8p11.21) (Tables II, III) [26]. Missense mutations in position p.R344C (22%) and p.S518F (29%) [27] are most frequently observed. MPS IIIC can be associated with other diseases such as Retinitis pigmentosa, when HGSNAT missense mutations p.G262R and p.S539C in membrane domain of the enzyme are present [28–30].
Mucopolysaccharidosis IIID
Mucopolysaccharidosis IIID is caused by GNS gene (12q14.3) mutations (~5% of MPS III, Tables II, III), which encodes lysosome N-acetylglucosamine-6-sulfatase. This enzyme contain 552 amino acids and 13 glucosylation (modification) sites [31]. Missense mutation occur in 13% of all MPS IIID cases.
Mucopolysaccharidosis IIIE
Mutations in ARSG gene (17q24.2) cause MPS IIIE (~2% of MPS-III, Tables II, III). The gene contains eleven exon, the protein N-glucosamine 3-O-sulfatse consist of 525 amino acids and four glucosylation (modification) sites in asparagine [32–4].
Table III
Summary of the MPS III associated with other diseases and gene mutation
Laboratory diagnostic methods for mucopolysaccharidosis
Stepwise diagnostics of MPS is done biochemically, for example by detection of heparan sulfate levels in urine of patients. Dimethylmethylen blue is used for heparan sulfate detection at the wavelength of 520 nm in a spectrophotometer [35, 36]. Another method is high-resolution cellulose acetate electrophoresis of a urine sample which can separate and identify heparan sulfate [37]. Finally, tandem mass spectrometry can be applied for diagnosis of MPS [38]; identification of specific oligosaccharides that are accumulate as a results of enzyme deficiency can be picked up by that [38–40]. Recently it was also suggested to measure protein amounts involved in MPS from dried blood spot using multiplex immune–quantification assay [41, 42].
The method to identify the underlying genetic cause and specific subtype of a suggested MPS is genomic DNA sequencing (NGS-next generation sequencing); also carrier status of parents can be assessed by this [43].
Postnatally, urine or peripheral blood can be studied. Prenatal genetic diagnostic is possible after invasive prenatal diagnostics, as chorionic villus sampling, amniocentesis or umbilical cord blood sampling [44].
Therapy for mucopolysaccharidosis patients
Most of the following treatment options are still experimental.
Enzyme replacement therapy
Enzyme replacement therapy is a possible approach to treat MPS patients. Injection of human sulfaminidase directly in brain [45] or cerebrospinal fluid via the cerebellomedullary cistern [46] has been shown to be principally possible in MPS IIIA mice [47] (Fig. 2A).
Figure 2
Potential therapy attempts to treat MPS. Schematic representations of: A) enzyme replacement therapy (ERT) to replace mutated form of protein; B) substrate reduction therapy (SRT), reduction of storage of undegraded substrate; C) pharmacological chaperone to correct substrate miss-folding; D) stem cell therapy (SCT) used for replacement of diseased cells; E) gene therapy to provide correct form of a mutated gene
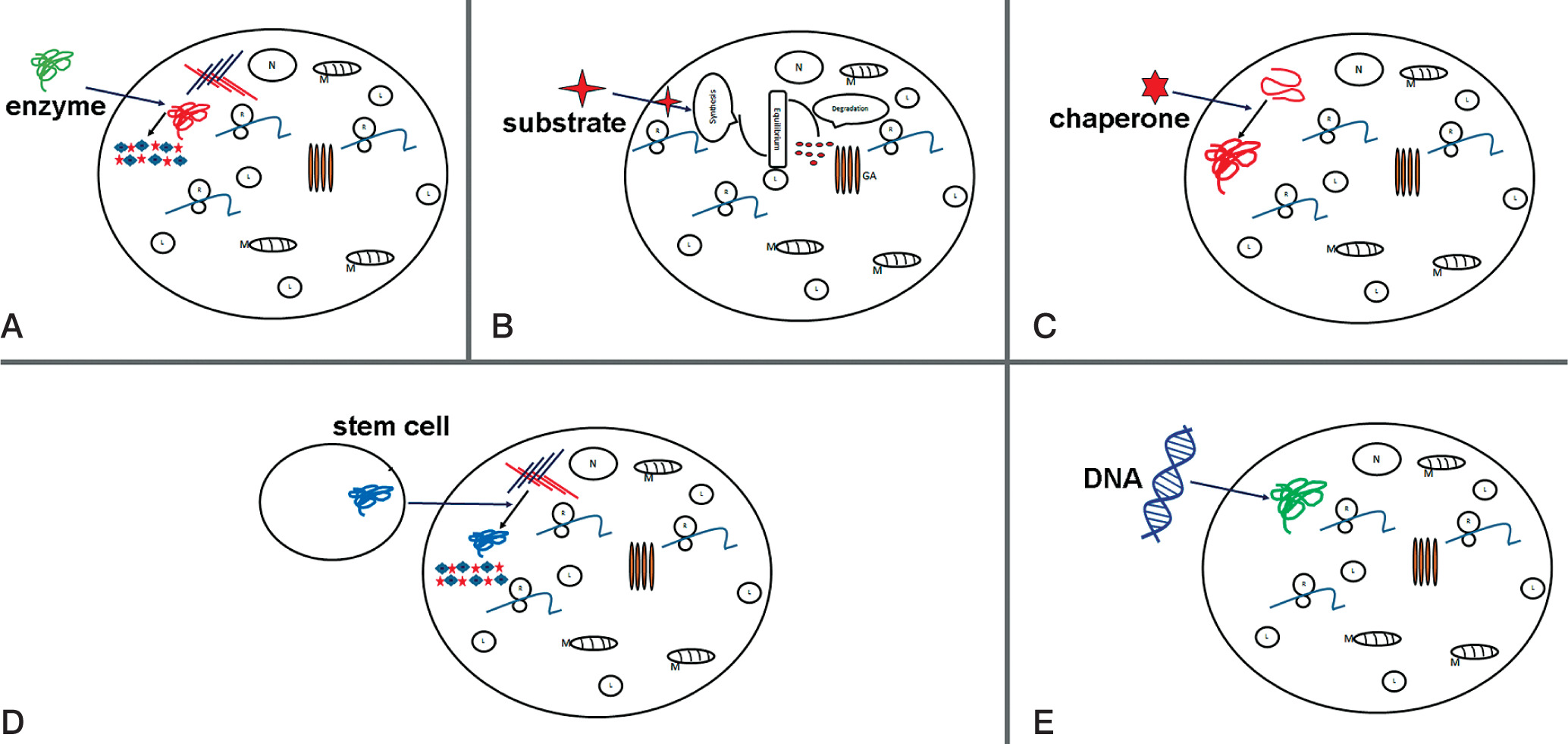
Substrate reduction therapy
Substrate reduction therapy (STR) in another alternative therapy approach and outline in Figure 1B. Aim is here to approach molecular targets decreasing production of accumulated substrate and to restore balance between synthesis and degradation. This method is approved e.g. for treatment of lysosomal disorders with neurological and non-neurological symptoms [48]. For STR RNAi can be applied to key genes participating in GAG synthesis, such as EXTL other genes involved in linkage region formation. Dziedzic et al., applied siRNA to down-regulate XYLT1, XYTL2, GALTI and GALTII [49].
This strategy is successfully applied in treatment of MPS I and MPS IIIA, resulting in reduction of mRNA and protein levels of corresponding genes and a significant decrease in GAG synthesis. Using of shRNA (short hairpin RNA) has been shown to have a positive effect in downregulation of two other genes EXTL2 and EXTL3; both genes influence reduction of GAG synthesis [50] (Fig. 2B).
Pharmacological therapy
In many cases, mutation is shown to lead production of misfolded protein, which become rapidly degraded. The activation of chaperons (cellular proteins) may help functional correct protein folding. As shown in Fig. 2C, small compounds as actin-like chaperones (= amino and iminosugars) prevent protein misfolding by acting as enzyme inhibitors [51]. This approach is used to treat different LDS diseases such Farby, Morquio B, Pompe, Gaucher and Krabbe diseases [52].
The component is an isoflavine which is purified from soy and has the ability to reduce GAG (glucoaminglucans) level. The overall mechanism is not clarify. However, it seems likely that the mechanism involves protein kinase inhibitory, which in turn induces TFEB (transcription factor EB) transcriptional activity [53, 54].
Growth medium supplemented by genistein has been shown to reduce GAG level in skin fibroblast of MPS IIIA and MPS IIIB patients [55]. In murine model such treatment led to reduction of GAG storage [56]. Moreover, in a pilot study in patients with MPS IIIA and B administration of genistein reduced GAG levels in urine [57]. In mouse reduction of GAG levels was demonstrated after treatment with rhodamine in the liver and the brain [58].
Stem cell therapy
Stem cell therapy was suggested for treatment of neurological diseases in order to produce correct forms of enzyme (Figure 2D). Bone marrow transplantation can be used for treatment of neurological diseases, but in MPS III diseases this approach was not successful [59]. The treatment with hematopoietic stem cell in patients show that in brain cells microglia can be replaced and become enzyme secretion donor cells [60]. Recently genetic modification of hematopoietic stem cells carrying normal copies of the SGSH or NAGLU genes led in mouse model to enzyme production [61–64].
iPSC (induced pluripotent stem cell) development technology gave new possibilities for generation of neural stem cell (NSC). After transplantation of NSC they could migrate over long distances within brain and were integrated in host network. This form of treatment recovered neurological pathology of MPS VII [65] and MPS IIIB [66].
Gene therapy
The gene therapy is used to enlargement ERT (enzyme replacement therapy) (Fig. 1E) so far is attempted to introduce the coding sequence of the protein. The cell manipulation will possess in enzyme activity but also will participate in enzyme secretion and circulation up to altered cell. The sulfamidase injection as a recombinant adeno-associated virus (AAV) will improve coding sequence and in this form will increase enzyme activity (sulfamidase) in brain of mice [67]. This strategy now is used in human clinical traits [68].
Gene therapy is most promising therapeutic option for treatment of MPS diseases, and 5–15% of enzyme activity can be recovered in affected patients [69]. Several viral vectors are used for treatment of MPS patients, such as retroviruses, lentiviruses, adenoviruses and adeno-associated viruses (AAV). Using of nonviral vector also led to increased enzyme activity and GAG reduction in lysosome [70]. Adeno-associated viruses treatment seems to be suited best for gene therapy in neurological disorders. AAV therapy could e.g. induce cell death in murine neural cells and hippocampus, suggesting that these approaches should be carefully evaluated [71]. AVV therapy and other viral gene therapies may be associated with significant side effects, particularly during development. Adeno-associated virus serotype 5 (AAV5) has been used for treatment of MPS IIIB patients, results shown improvement of neurological progression in patients inducing sustained enzyme production in the brain [72]. AAVrh10 (adeno-associated virus serotype rh10) was used to integrate intact SGSH gene in MPS IIIA model mice. This treatment reduced HS accumulation and microglia activity administration [73]. The AAV8 delivered effectively NAGLU gene in MPS IIIB animal model and also facilitated robust somatic transduction of the heart and liver [74].
Conclusions
In this review we provide an overview for molecular basis of MPS III, laboratory diagnostic methods such biochemical detection of heparan sulfate in patients with MPS III. Genes responsible for MPS III have been identified by characterizing the functional role of gene products in the metabolism. A number of genetic and biochemically methods have been adopted in laboratory for diagnosis. Although four types of MPS III diagnosis remain difficult, our recommendations for screening and diagnosis are as follows:
All patients with speech delay, deficit or hyperactivity disorders, autism should be screened for MPS III. DMB (1,2-diamino-4,5-methylenedioxybenzene-2HCl) assay is strongly recommended.
All positive samples from quantitative assay should be investigate by electrophoresis, GAG depolymerization followed by HPLC/MS/MS analysis.
An enzyme activity assay must be done to confirm the diagnosis.
Molecular genetic testing should be offered to all patients; this test is informative for the family when they making decision for family plan.
Therapeutic options for MPS III disease, once considered untreatable, are available for patients with these diseases. The iPSC have been established, and will be very useful in drug screening studies to identify the drugs which will be potential for human treatment.